NUEVOS REARREGLOS ESTRUCTURALES EN LEUCEMIA LINFOCITICA CRONICA/LINFOMA DE LINFOCITOS PEQUEÑOS
(especial para SIIC © Derechos reservados)
Coautores
Christian Pablo Chena* Irma Rosa Slavutsky**
Licenciado en Genética. Departamento de Genética, Instituto de Investigaciones Hematológicas “Mariano R. Castex”, Academia Nacional de Medicina.*
Doctora en Medicina. Departamento de Genética, Instituto de Investigaciones Hematológicas “Mariano R. Castex”, Academia Nacional de Medicina.**
Recepción del artículo: 11 de enero, 2005
Aprobación: 14 de febrero, 2005
 Conclusión breve
El trabajo informa sobre cuatro anomalías estructurales nuevas, una de ellas recurrente, y sugiere que éstas podrían implicar eventos genéticos asociadas a enfermedad inestable.
Resumen
La leucemia linfocítica crónica/linfoma de linfocitos pequeños (LLC/LLP) presenta una evolución clínica muy variable, encontrándose rearreglos genómicos asociados con diferentes grupos de riesgo. Este trabajo se centra en el análisis de rearreglos estructurales de los cromosomas 2, 12 y 17; se describen 4 anomalías nuevas, detectadas en un total de 92 pacientes. Simultáneamente, se efectuó una exhaustiva revisión de la literatura. Se efectuó estudio citogenético e hibridación in situ con fluorescencia (FISH). Cuatro casos mostraron anomalías que afectaban el cromosoma 17, siendo el cromosoma 2, con punto de ruptura en 2p21, el cromosoma partner más frecuente. Un paciente presentó coexistencia de LLC/LLP con enfermedad de Hodgkin, subtipo celularidad mixta, detectándose el marcador psu dic (17;2)(p112;p21), con deleción monoalélica del gen TP53. Dos casos mostraron una nueva anomalía recurrente: t(2;17)(p21;q23), asociada a evolución clonal en ambos pacientes y a deleción 11q23 en uno de ellos. Las restantes alteraciones fueron el der(17)t(12;17)(q13;q25) y una translocación compleja t(5;12;19)(q15;p11;q13). Cuatro pacientes tuvieron curso clínico adverso y murieron debido a progresión de su enfermedad. Nuestro trabajo aporta cuatro anomalías estructurales nuevas, una de ellas recurrente, y sugiere que éstas podrían implicar eventos genéticos asociadas a enfermedad inestable.
Palabras clave
LLC/LLP, enfermedad de Hodgkin, citogenética, FISH
Clasificación en siicsalud
Conclusión breve
El trabajo informa sobre cuatro anomalías estructurales nuevas, una de ellas recurrente, y sugiere que éstas podrían implicar eventos genéticos asociadas a enfermedad inestable.
Resumen
La leucemia linfocítica crónica/linfoma de linfocitos pequeños (LLC/LLP) presenta una evolución clínica muy variable, encontrándose rearreglos genómicos asociados con diferentes grupos de riesgo. Este trabajo se centra en el análisis de rearreglos estructurales de los cromosomas 2, 12 y 17; se describen 4 anomalías nuevas, detectadas en un total de 92 pacientes. Simultáneamente, se efectuó una exhaustiva revisión de la literatura. Se efectuó estudio citogenético e hibridación in situ con fluorescencia (FISH). Cuatro casos mostraron anomalías que afectaban el cromosoma 17, siendo el cromosoma 2, con punto de ruptura en 2p21, el cromosoma partner más frecuente. Un paciente presentó coexistencia de LLC/LLP con enfermedad de Hodgkin, subtipo celularidad mixta, detectándose el marcador psu dic (17;2)(p112;p21), con deleción monoalélica del gen TP53. Dos casos mostraron una nueva anomalía recurrente: t(2;17)(p21;q23), asociada a evolución clonal en ambos pacientes y a deleción 11q23 en uno de ellos. Las restantes alteraciones fueron el der(17)t(12;17)(q13;q25) y una translocación compleja t(5;12;19)(q15;p11;q13). Cuatro pacientes tuvieron curso clínico adverso y murieron debido a progresión de su enfermedad. Nuestro trabajo aporta cuatro anomalías estructurales nuevas, una de ellas recurrente, y sugiere que éstas podrían implicar eventos genéticos asociadas a enfermedad inestable.
Palabras clave
LLC/LLP, enfermedad de Hodgkin, citogenética, FISH
Clasificación en siicsalud
Artículos originales> Expertos del Mundo>
página www.siicsalud.com/des/expertos.php/71972
Especialidades
 Principal: Genética Humana, Hematología,
Principal: Genética Humana, Hematología,
Relacionadas: Diagnóstico por Laboratorio, Medicina Interna, Oncología,
Enviar correspondencia a:
Roxana Cerretini. Departamento de Genética, Instituto de Investigaciones Hematológicas, Academia Nacional de Medicina. Pacheco de Melo 3081 (1425) Buenos Aires, Argentina
Patrocinio y reconocimiento
Agradecimientos: Este trabajo fue realizado con subsidios otorgados por el Consejo Nacional de Investigaciones Científicas y Técnicas (CONICET), la Agencia Nacional de Promoción Científica y Técnica (ANPCyT), Fundación “Alberto J. Roemmers” y Fundación Acción Oncohematológica.
NOVEL STRUCTURAL ABERRATIONS IN CHRONIC LYMPHOCYTIC LEUKEMIA/SMALL LYMPHOCYTIC LYMPHOMA
Abstract
Chronic lymphocytic Leukemia/small lymphocytic lymphoma (CLL/SLL) is a lymphoproliferative disorder characterized by a highly variable clinical course and specific genetic alterations have been associated with different risk groups. In the present study, we focused on the analysis of structural anomalies of chromosomes 2, 12 and 17. In addition, a exhaustive revision of the literature was performed. We identified four new genomic rearrangements detected among 92 evaluated patients. Cytogenetic studies and fluorescence in situ hybridization (FISH) analysis were performed. Four cases showed chromosome 17 structural anomalies and chromosome 2 with breakpoint at p21 was the most frequent chromosome partner. A psu dic (17;2)(p11.2;p21), leading to a TP53 deletion, was observed in a patient who developed a mixed cellularity Hodgkin’s disease coexisting with the CLL/SLL. Two cases had a new recurrent translocation t(2;17)(p21;q23), both associated with clonal evolution and one of them also with a 11q23 deletion. In addition, a der(17)t(12;17)(q13;q25) and a complex translocation t(5;12;19) (q15;p11;q13) were also found. Four patients presented an adverse clinical outcome and died due to disease progression. These uncommon abnormalities may have implications for the understanding of genetic events associated with evolving disease.
Key words
CLL/SLL, Hodgkin’s disease, cytogenetic, FISH
NUEVOS REARREGLOS ESTRUCTURALES EN LEUCEMIA LINFOCITICA CRONICA/LINFOMA DE LINFOCITOS PEQUEÑOS
(especial para SIIC © Derechos reservados)
Artículo completo
Introducción
La leucemia linfocítica crónica/linfoma de linfocitos pequeños (LLC/LLP) es un proceso linfoproliferativo crónico, con una incidencia que corresponde al 7% del total de los linfomas no Hodgkin (LNH),1 cuya patogénesis, así como su célula de origen no están aún bien definidas.2,3 Presenta un curso clínico muy variable, situación que determina que cada paciente tenga un riesgo individual de evolución de la enfermedad y de supervivencia. Esta marcada heterogeneidad en términos de morfología, inmunofenotipo y evolución clínica ha orientado la investigación hacia la búsqueda de factores pronósticos robustos de utilidad en la valoración de este riesgo. El estado mutacional de la región variable de los genes de las cadenas pesadas de las inmunoglobulinas (IgVH),4 es uno de los marcadores que mejor correlación tiene con la evolución clínica,5,6 con la salvedad de que los métodos requeridos para tal valoración no son simples ni están disponibles, en la mayoría de los casos, en la práctica clínica. La expresión de ZAP-70, considerado como el subrogante del estado mutacional de IgVH, constituye un buen parámetro, que puede ser fácilmente detectable por citometría de flujo, ya que presenta buena asociación con progresión de la enfermedad y supervivencia.7 Contrariamente, el valor de CD38 como factor pronóstico es controvertido,8,9 sobre todo si se tiene en cuenta que sus niveles de expresión pueden variar durante el curso de la enfermedad.10
Simultáneamente, las anomalías cromosómicas tienen un papel relevante en la definición de diferentes grupos de riesgo para esta entidad, habiéndose encontrando buena correlación entre las características clínico-biológicas y los patrones citogenéticos.11-13 Los estudios de citogenética convencional demuestran que 40% a 50% de los casos con LLC/LLP presentan alteraciones cromosómicas clonales,14,15 entre las que la trisomía 12 es la anomalía numérica más comúnmente descrita (7% a 25%), seguida por alteraciones estructurales de los brazos largos de los cromosomas 13 (20%) y 14 (4%).15-17 No obstante, con la introducción de las técnicas citomoleculares se incrementa ampliamente la sensibilidad del análisis citogenético, se detectan alteraciones en el 80% de los casos, y se modifica además el orden de incidencia de las alteraciones: deleción 13q14 (50%-60%), deleción 11q22-q23 (20%-40%), trisomía 12 (10%-> 30%), pérdida monoalélica de 17p13 (7%-17%), lugar donde se ubica el gen TP53, y deleciones en 6q (6%). Aproximadamente el 20% de los pacientes presentan cariotipo normal, sin los rearreglos genómicos descritos con anterioridad.18
Asimismo, se sabe que los casos de LLC/LLP con deleción en 13q14 sola tienen mejor pronóstico que los pacientes con cariotipo normal, la presencia de trisomía 12 se encuentra asociada con pronóstico intermedio, en tanto que las deleciones o los rearreglos, o ambos, que involucren 11q22-q23 y 17p13 muestran en común un curso clínico adverso y corta supervivencia.11,18,19
Si bien se conocen los rearreglos genómicos más frecuentes no es claro aún el proceso de desregulación génica que da origen a la LLC/LLP, por lo que resulta de importancia dirigir las investigaciones hacia la búsqueda de nuevas alteraciones que permitan establecer asociaciones más precisas entre las anomalías cromosómicas y las características clínico-biológicas de esta patología. En el presente trabajo, centramos nuestro interés en el análisis de los rearreglos estructurales de los cromosomas 2, 12 y 17, se describien 4 anomalías cromosómicas nuevas y se efectúa una exhaustiva revisión de la literatura.
Materiales y métodos
En nuestra institución fueron estudiados 92 pacientes con diagnóstico de LLC/LLP durante el período 1993-2003. El 5.3% (5/92) de los casos (cuatro varones y una mujer; edad media 59 años; intervalo 50-70 años) presentaron rearreglos estructurales nuevos que involucraban los cromosomas 2, 12 y 17. La Tabla 1 detalla edad, sexo, tipo de muestra analizada, respuesta al tratamiento y supervivencia correspondientes a cada caso. Sus características hematológicas fueron previamente descritas.20

Dos pacientes (Casos 2 y 5) presentaron perfiles clínicos, inmunofenotípicos e histopatológicos compatibles con LLP,21,22 en tanto que los tres casos restantes (Casos 1, 3 y 4) reunieron los criterios para LLC.23 El Caso 1 fue un linfoma compositum que desarrolló enfermedad de Hodgkin (subtipo: celularidad mixta) 19 meses después del diagnóstico de LLC, mostrando el perfil inmunofenotípico característico de linfocitos B y de células de Reed-Stemberg (RS).
Los estudios citogenéticos y de hibridación in situ con fluorescencia (FISH) fueron realizados al diagnóstico o previamente al tratamiento en los Casos 2, 3 y 4; luego de varios esquemas quimioterapéuticos, en el Caso 5, y al diagnóstico y 19 meses después, en el Caso 1. El 80% (4/5) de los pacientes habían fallecido al momento de este estudio.
El análisis cromosómico fue realizado en cultivos de corto término (24-48 hs) de médula ósea (MO) o biopsia de nódulo linfático (BNL) o ambos, en medio F-10 suplementado con suero fetal bovino al 15%, empleándose bandeo GTG (bandas G con tripsina y coloración con Giemsa) y la nomenclatura ISCN24 para la identificación y descripción de las anomalías cromosómicas. Este estudio se complementó con FISH mediante el empleo de sondas del pintado total de los cromosomas, centroméricas y específicas para locus, siguiendo los protocolos estándar.
Resultados
La Tabla 1 muestra los estudios citogenéticos y citomoleculares correspondientes a los cinco pacientes estudiados. Cuatro casos presentaron rearreglos estructurales del cromosoma 17 y un caso una translocación compleja que involucraba al cromosoma 12.
El Caso 1 presentó un cromosoma seudodicéntrico psu dic(17;2)(p11.2;p21) como única anomalía citogenética. Asimismo, los estudios de hibridación in situ revelaron la presencia del virus de Epstein Barr (EBV) en las células de RS, en tanto que las células de la LLC/LLP fueron negativas.
Los Casos 2 y 3 mostraron la misma translocación t(2;17)(p21q23), asociada a trisomías 12 y 17 y monosomía 10 (Caso 2) y a del(11)(q23) (Caso 3).
Los Casos 4 y 5 presentaron cariotipos complejos que incluyeron el der(17)t(12;17)(q13;q25) (Caso 4) y un rearreglo complejo t(5;12;19)(q15;p11;q13) (Caso 5).
Discusión
En este estudio se describen 5 pacientes con LLC/LLP que presentaron rearreglos cromosómicos estructurales no publicados previamente en la literatura. El cromosoma 17 estuvo involucrado en cuatro de ellos, en 3 de los cuales el cromosoma 2 fue el cromosoma partner, mientras que el caso restante presentó una translocación compleja que comprometía el brazo corto del cromosoma 12.
En el Caso 1 se observó un cromosoma seudodicéntrico: psu dic (17;2)(p11.2;p21) como única anomalía, dando origen a una deleción monoalélica del gen TP53. Es interesante remarcar que en las neoplasias hematológicas los cromosomas seudodicéntricos se observan con menor frecuencia que los cromosomas dicéntricos y, en particular, su hallazgo es un evento raro en LLC/LLP, siendo el dic(17;18)(p11;p11) o der(17;18)(q10;q10) el más frecuente en esta patología, con 11 casos publicados.25 Específicamente, la participación de los cromosomas 2 y 17 en la formación de este tipo de aberraciones es sumamente infrecuente, se encontró una sola referencia previa en la literatura, con diferentes puntos de ruptura que los observados en nuestro paciente26 (Tabla 2). Distintos trabajos mostraron que las deleciones del gen TP53 tiene fuertes implicancias en el curso clínico de la enfermedad, con una supervivencia media significativamente más corta, estadio clínico avanzado y resistencia a la quimioterapia.18,19 Estos hallazgos están en concordancia con nuestro caso, que mostró progresión de la enfermedad y una clara falta de respuesta a los diferentes esquemas quimioterapéuticos empleados (agentes alquilantes, antraciclinas y análogos de las purinas). Además, 19 meses después del diagnóstico inicial de LLC/LLP, la paciente contrajo enfermedad de Hodgkin (EH) con positividad para EBV. Se sabe que aproximadamente el 3% al 10% de los casos de LLC/LLP evolucionan a un linfoma B difuso de células grandes (LBDCG), proceso conocido como síndrome de Richter.27 En un pequeño número de casos se produce una transformación a un componente con características de EH con el fenotipo característico de las células de RS.28-30 Es interesante destacar que los pacientes con LLC/LLP tienen un riesgo 8 veces mayor de desarrollar EH,31 que es una de las neoplasias secundarias más frecuentes en esta entidad. Existen evidencias de que las células de RS representan poblaciones clonales de linfocitos B derivadas del centro germinal. En muy pocos casos, el análisis de clonalidad de las células correspondientes a LLC y a EH resultaron en la detección del mismo rearreglo clonal del gen de la cadena pesada de las inmunoglobulinas, indicando que ambas derivaban de la misma célula B precursora. Esta situación sugeriría la presencia de eventos moleculares diferentes en el precursor común que llevarían a la aparición de dos poblaciones celulares con capacidad de expansión clonal. Sin embargo, en un alto porcentaje de casos, las células de RS positivas para el EBV no están genéticamente relacionadas con el clon tumoral de la LLC, lo que indica que el EBV podría tener un papel importante en el desarrollo de la EH, situación que es sugerida para nuestro paciente.32-34
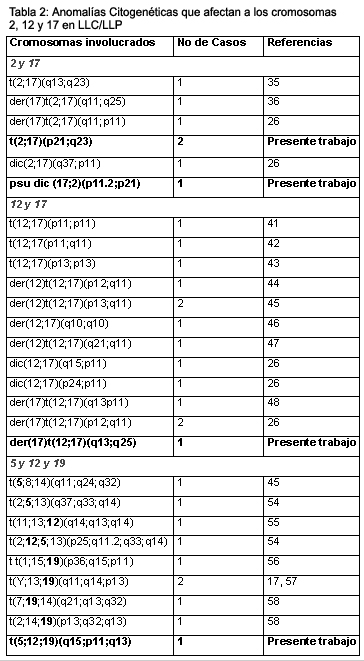
Tres casos presentaron anomalías del brazo largo del cromosoma 17. Dos pacientes mostraron la t(2;17)(p21;q23), la cual podría representar una nueva anormalidad cromosómica recurrente en LLC/LLP. Uno de ellos se encontraba fallecido al momento de este estudio, en tanto que el otro paciente está vivo y en remisión parcial. Es interesante remarcar que éste fue el único rearreglo balanceado del cromosoma 17 observado en nuestra serie. Este hallazgo resulta de importancia, no sólo por la recurrencia, sino porque las translocaciones balanceadas son, en general, anomalías muy poco frecuentes en esta patología y usualmente involucran alguno de los genes de inmunoglobulinas.25 En particular, sólo han sido publicadas hasta el presente, tres translocaciones que involucrando los cromosomas 2 y 17, una balanceada y dos desbalanceadas, ninguna recurrente26,35,36 (Tabla 2). Si bien la LLC/LLP es considerada una enfermedad genéticamente estable, diferentes estudios indicarían que la evolución clonal no es infrecuente en estos pacientes y que estaría asociada con enfermedad progresiva o avanzada.37 Ambos pacientes mostraron evolución cariotípica y el Caso 3 mostró deleción de 11q23, región donde se localiza el gen ATM. Dicha deleción es considerada como una de las alteraciones cromosómicas más comunes, después de las aberraciones de 13q, encontrándose asociada con enfermedad agresiva con cariotipo inestable. Estos pacientes tienen un cuadro clínico característico, con presentación de la enfermedad a una edad más temprana, con estadios avanzados y linfoadenopatías extensas.11,18,37 Un estudio reciente38 muestra que esta anomalía podría ser adquirida durante el curso de la enfermedad, algunas veces se la encuentra asociada con una progresión clínica abrupta. En coincidencia con estos datos, al momento de nuestro primer análisis, el paciente se encontraba con progresión de la enfermedad, caracterizada por cortos intervalos libres de tratamiento, linfocitosis progresiva y aumento de las adenopatías, luego de 4 años de enfermedad estable.
Este estudio también muestra que el cromosoma 2 con punto de ruptura en p21 es el cromosoma partner más frecuentemente involucrado en rearreglos de ambos brazos del cromosoma 17, generando tanto anomalías simples como dicéntricas. En este aspecto la literatura documenta que los cromosomas 2, 4, 8, 12, 13, 14, 15, 18, 19, 20 y 22 son los más comúnmente implicados en rearreglos del cromosoma 17, con baja frecuencia de dicéntricos.25 Las bandas 2p21-22 se encuentran frecuentemente rearregladas en pacientes con leucemias y neoplasias linfoides,25 región donde, es de destacar, fueron mapeados el gen supresor de tumor de la proteína quinasa activada por RNA doble cadena (PKR)39 y el gen MSH2 de reparación de errores de apareamiento.40
El caso restante con anomalía de 17q presentó una translocación desbalanceada: der(17)t(12;17)(q13;q25), como parte de un cariotipo complejo. Este paciente tuvo un curso clínico adverso y murió debido a progresión de la enfermedad. En la literatura hay descritos once rearreglos que involucran ambos cromosomas (Tabla 2), de los cuales 3 son translocaciones balanceadas sin recurrencia,41-43 y 8, rearreglos desbalanceados, dos de ellos recurrentes20,26,44-48 (Tabla 2). De los tres pacientes con translocaciones que comprendían el brazo largo del cromosoma 17 (Casos 2, 3 y 4), dos mostraron el mismo punto de ruptura en la banda 17q23 y el restante en la banda 17q25. Las anomalías del brazo largo de dicho cromosoma no son frecuentes en pacientes con LLC/LLP, no obstante, el punto de ruptura 17q25 se observa más frecuentemente involucrado que la banda 17q23.25 Por otra parte, una revisión de la literatura (Tabla 2), muestra que en los rearreglos (12;17), el punto de ruptura 17q11 es el más frecuentemente afectado. Recientemente, el gen RAC3 de la guanosina trifosfatasa (GTPasa) fue mapeado en 17q25.3, se reconoce actualmente que las proteínas RAC constituyen un componente necesario de la transformación celular mediada por RAS y relevante para la patogénesis de una amplia variedad de neoplasias.40,49 Asimismo, en 17q25.1 se ubica el gen DMC1 (downregulated in multiple cancer-1) que produce una proteína integral de membrana, cuya pérdida de expresión se observa en muchas neoplasias.50 Por otro lado, las aberraciones estructurales del brazo largo del cromosoma 12 fueron observadas en alrededor del 5% de los casos de LLC/LLP.14 Varios genes han sido identificados en la banda 12q13, incluyendo MDM2 (mouse double minute 2), que codifica para una proteína de 90-kD que puede unirse e inactivar la proteína p53, promoviendo el desarrollo tumoral. Diferentes estudios documentaron que dicho gen se encuentra sobrexpresado o rearreglado en pacientes con LLC.51,52 Asimismo, se informó un caso con diagnóstico de LLC típica y t(6;12) resultando en una ruptura en 12q13, lo que sugiere que esta banda portaría un gen específico que, al alterarse, contribuiría a la aparición de esta patología.53
El Caso 5 presentó una translocación compleja t(5;12;19) no descrita previamente. Las translocaciones complejas son rearreglos muy poco frecuentes en este subtipo histológico y corresponden al 2.4% de los casos.25 Particularmente, fueron descritos 8 casos de LLC/LLP con translocaciones complejas que involucran algunos de los tres cromosomas implicados en nuestro paciente (Tabla 2).17,45,54-58 Los rearreglos del brazo corto del cromosoma 12 son comunes en una amplia variedad de neoplasias hematológicas, particularmente síndromes mielodisplásicos y leucemias agudas. Por el contrario, estos rearreglos constituyen eventos raros en pacientes con LLC/LLP.25 Recientemente se ha propuesto que las aberraciones de 12p podrían ser anomalías primarias en esta patología, y es interesante destacar que uno de los casos publicados con compromiso de 12p tuvo un curso clínico agresivo.59 En lo que respecta al cromosoma 19, sabemos que a nivel de 19q13, se localiza el oncogén BCL-3 (B cell lymphoma leukemia) que codifica una proteína de la familia I-κB.58,60 Esta actúa como coactivadora de los factores de transcripción NF-κB, involucrados en el control del ciclo celular y en la determinación del linaje, y cumple un papel importante en el desarrollo de las células B y en la activación de los linfocitos B y T. Nuestro caso fue refractario a la quimioterapia, y murió debido a una rápida progresión de la enfermedad, 9 meses después del diagnóstico de LLP.
En conclusión, se identificaron 4 rearreglos cromosómicos estructurales, no mencionados previamente en la literatura, de los cuales uno podría representar una nueva anomalía recurrente en LLC/LLP. Estas alteraciones citogenéticas se encontraron asociadas con curso clínico adverso, con enfermedad progresiva y corta supervivencia, lo cual puede ser de importancia en la comprensión de los eventos genéticos asociados con progresión de la enfermedad.
Los autores no manifiestan “conflictos de interés”.
Bibliografía del artículo
- NHLCP: The Non-Hodgkin´s lymphoma Classification Project. A clinical evaluation of the International Lymphoma Study Group. Classification of Non-Hodgkin´s lymphoma. Blood 1997; 89: 3909-18.
- Klein U, Tu Y, Stolovitzky GA y col. Gene expression profiling of B cell chronic lymphocytic leukemia reveals a homogeneous phenotype related to memory B cells. J Exp Med. 2001, 194:1625-38.
- Rosenwald A, Alizadeh AA, Widhopf G y col. Relation of gene expression phenotype to immunoglobulin mutation genotype in B cell chronic lymphocytic leukemia. J Exp Med. 2001; 194:1639-47.
- Stilgenbauer S, Bullinger L, Lichter P y col. Study Group (GCLLSG). Chronic lymphocytic leukemia. Genetics of chronic lymphocytic leukemia: genomic aberrations and V(H) gene mutation status in pathogenesis and clinical course. Leukemia. 2002; 6:993-1007.
- Hamblin TJ, Davis Z, Gardiner A y col. Unmutated IgV genes are associated with a more aggressive form of chronic lymphocytic leukemia. Blood 1999; 94: 1848-54.
- Damle RN, Wasil T, Fais F y col. Ig V gene mutation status and CD38 expression as novel prognostic indicators in chronic lymphocytic leukemia. Blood. 1999; 94:1840-7.
- Crespo M, Bosch F, Villamor N y col. ZAP-70 expression as a surrogate for immunoglobulin-variable-region mutations in chronic lymphocytic leukemia. N Engl J Med. 2003; 348:1764-75.
- Del Poeta G, Maurillo L, Venditti A y col. Clinical significance of CD38 expression in chronic lymphocytic leukemia. Blood. 2001; 98: 2633-9.
- Ibrahim S, Keating M, Do KA y col. CD38 expression as an important prognostic factor in B-cell chronic lymphocytic leukemia. Blood. 2001; 98:181-6.
- Hamblin TJ, Orchard JA, Gardiner A y col. Immunoglobulin V genes and CD38 expression in CLL. Blood. 2000; 95: 2455-7.
- Döhner H, Stilgenbauer S, James MR y col. 11q deletions identify a new subset of B-cell chronic lymphocytic leukaemia characterized by extensive nodal involvement and inferior prognosis. Blood 1997; 89: 2516-22.
- Cuneo A, Bigoni R, Castoldi G. Towards a clinically relevant cytogenetic classification of chronic lymphocytic leukemia and related disorders. Haematologica. 1998; 83: 577-9.
- Oscier DG. Cytogenetics and molecular genetics of chronic lymphocytic leukaemia. Haematologica 1999; 84 Suppl EHA-4: 88-91.
- Juliusson G, Oscier DS, Fitchett M y col. Prognostic subgroups in B-cell chronic lymphocytic leukemia defined by specific chromosomal abnormalities. N Engl J Med 1990; 323: 720-24.
- Juliusson G, Oscier D, Gahrton G. Cytogenetic findings and survival in B-cell chronic lymphocytic leukemia. Second International Working Party on Chromosomes in CLL (IWCCLL)IWCCLL compilation of data on 662 patients. Leuk Lymphoma 1991; 5: 21-25.
- Juliusson G, Gahrton G. Chromosome aberrations in B-cell chronic lymphocytic leukemia: Pathogenetic and clinical implications. Cancer Genet. Cytogenet 1990; 45: 143-60.
- Peterson LC, Lindquist LL, Church S y col. Frequent clonal abnormalities of chromosome band 13q14 in B-cell chronic lymphocytic leukemia: multiple clones, subclones, and nonclonal alterations in 82 midwestern patients. Genes Chrom Cancer 1992; 4: 273-80.
- Döhner H, Stilgenbauer S, Benner A y col. Genomic aberrations and survival in chronic lymphocytic leukemia. N Eng J Med 2000; 343: 1910-6.
- Döhner H, Fischer K, Bentz M y col. p53 gene deletion predicts for poor survival and non-response to therapy with purine analogous in chronic B-cell leukemias. Blood 1995; 85: 1580-9.
- Cerretini R, Chena C, Giere I y col. Structural aberrations of chromosomes 17 and 12 in chronic B cell-disorders. Eur J Haematol 2003: 71: 433-38.
- Harris NL, Jaffe ES, Stein H. A revised European-American classification of lymphoid neoplasms a proposal from the Internacional Lymphoma study group. Blood 1994;84: 1361-92.
- Harris NL, Jaffe ES, Diebold J. World Heath Organization classification of neoplastic diseases of the hematopoietic and lymphoid tissues. Report of the clinical advisory Committee meeting-Airlie House, Virginia, November 1997. J Clin Oncol 1999; 17:3835-49.
- Cheson BD, Bennet JM, Grever M y col. National Cancer Institute sponsored. Working Group guidelines for chronic lymphocytic leukemia: revised guidelines for diagnosis and treatment. Blood 1996; 87: 4990-7.
- ISCN. An International System for Human Cytogenetic Nomenclature. Mitelman F, ed. Karger, Basel, 1995.
- Mitelman Database of Catalogue of Chromosome Aberrations in Cancer. 2004. Mitelman F, Johansson B, Mertens F, eds http://cgap.nci.nih.gov/Chromosomes/Mitelman.
- Callet-Bauchu E, Salles G, Gazzo S y col. Translocations involving the short arm of chromosome 17 in chronic B-lymphoid disorders: frequent occurrence of dicentric rearrangements and possible association with adverse outcome. Leukemia 1999; 13:460-8.
- Giles FJ, O´Brien SM, Keating MJ. Chronic lymphocytic leukemia in Richter´s transformation. Semin Oncol 1998; 25: 117-25.
- Choi H, Keller RH. Coexistence of chronic lymphocytic leukemia and Hodgkin´s disease. Cancer 1981; 48: 48-57.
- Butts C, Drouin J, Taylor R y col. Hodgkin´s disease in CLL. Am J Hematol 1995; 48: 134-5.
- Serratrice de Roux C, Coso D, Bouabdallah R y col. Chronic lymphocytic leukemia and Hodgkin´s disease. Clinicopathologic study of three cases with good prognosis. Haematologica 2000; 85: 878-9.
- Travis LB, Curtis RE, Hankey BF y col. Second cancers in patients with chronic lymphocybtic leukemia. J Natl Can Inst 1992; 84: 1422-7.
- Ohno T, Smir BN, Weisenburger DD y col. Origin of the Hodgkin/Reed-Sternberg cells in chronic lymphocytic leukemia with “Hodgkin´s transformation”. Blood 1998; 91: 1757-61.
- Kanzler H, Küppers R, Helmes S y col. Hodgkin and Reed-Sternberg-like cells in B-cell chronic lymphocytic leukemia represent the outgrowth of single germinal –center B-cell-derived clones: potential precursors of Hodgkin and Reed-Sternberg cells in Hodgkin´s disease. Blood 2000; 95: 1023-31.
- Küppers R, Sousa AB, Baur AS y col. Common germinal-center B-cell origin of the malignant cells in two composite lymphomas, involving classical Hodgkin´s disease and either follicular lymphoma of B-CLL. Mol Med 2001; 7: 285-92.
- Stilgenbauer S, Dohner H, Bulgay-Morschel M y col. High frequency of monoallelic retinoblastoma gene deletion in B-cell chronic lymphoid leukemia shown by interphase cytogenetics. Blood. 1993; 81: 2118-24.
- Finn WG, Kay NE, Kroft SH y col. Secondary abnormalities of chromosome 6q in B-cell chronic lymphocytic leukemia: a sequential study of karyotypic instability in 51 patients. Am J Hematol 1998; 59 : 223-9.
- Fegan C, Robinson H, Thompson P y col. Karyotypic evolution in CLL: identification of a new sub-group of patients with deletions of 11q and advanced disease. Leukemia 1995; 9: 2003-8.
- Cuneo A, Bigoni R, Rigolin GM y col. Late appearance of the 11q22.3-23.1 deletion involving the ATM locus in B-cell chronic lymphocytic leukemia and related disorders. Clinico-biological significance. Haematologica 2002; 87: 44-51.
- Haus O. The genes of interferons and interferon-related factors: localization and relationships with chromosome aberrations in cancer. Arch Immunol Ther Exp 2000; 48: 95-100.
- Kotoula V, Hytiroglou P, Kaloutsi V y col. Mismatch repair gene expression in malignant lymphoproliferative disorders of B-cell origin. Leuk Lymphoma 2002; 43: 393-9.
- Fenaux P, Preudhomme C, Lai JL y col. Mutations of the p53 gene in B-cell chronic lymphocytic leukemia: a report on 39 cases with cytogenetic analysis. Leukemia 1992, 6: 246-50.
- Fleischman EW, Prigogina EL, Ilynskaya GW y col. Chromosomal characteristics of malignant lymphoma.Hum Genet 1989; 82: 343-8.
- Juliusson G, Friberg K, Gahrton G. Consistency of chromosomal aberrations in chronic B-lymphocytic leukemia. A longitudinal cytogenetic study of 41 patients. Cancer 1988; 62: 500-6.
- Dierlamamm J, Wlodarska I, Michaux L y col. FISH identifies different types of duplications with 12q13-15 as the commonly involved segment in B-cell lymphoproliferative malignancies characterized by partial trisomy 12. Genes Chrom Cancer 1997; 20: 155-66.
- Sato Y, Kobayashi H, Suto Y y col. Chromosomal instability in chromosome band 12p13: multiple breaks leading to complex rearrangements including cytogenetically undetectable sub-clones. Leukemia. 2001: 1193-202.
- Ravandi F, Hayes K, Cortes J y col. Translocation t(17;18)(q10;q10): a new nonrandom chromosomal translocation of clonal evolution in chronic myeloid leukemia. Cancer. 2001; 91: 1704-8.
- Bird ML, Ueshima Y, Rowley JD y col. Chromosome abnormalities in B cell chronic lymphocytic leukemia and their clinical correlations. Leukemia 1989, 82: 2943-47.
- Cuneo A, Balboni M, Piva N y col. Atypical chronic lymphocytic leukaemia with t(11;14)(q13;q32): karyotype evolution and prolymphocytic transformation.Br J Haematol. 1995; 90: 409-16.
- Morris CM, Haataja L, McDonald M y col. The small GTPase RAC3 gene is located within chromosome band 17q25.3 outside and telomeric of a region commonly deleted in breast and ovarian tumors. Cytogenet Cell Genet 2000; 89: 18-23.
- Harada H, Nagai H, Tsuneizumi M y col. Identification of DMC1, a novel gene in the TOC region on 17q25.1 that shows loss of expression in multiple human cancers. J Hum Genet. 2001; 46: 90-5.
- Huang YQ, Raphael B, Buchbinder A y col. Rearrangement and expression of MDM2 oncogene in chronic lymphocytic leukemia. Am J Hematol 1994; 47: 139-41.
- Haidar MA, El-Hajj H, Bueso-Ramos CE y col. Expression profile of MDM-2 proteins in chronic lymphocytic leukemia and their clinical relevance. Am J Hematol 1997; 54; 189-95.
- Stock AD, Dennis TR: A translocation breakpoint at chromosome band 12q13 associated with B-cell chronic lymphocytic leukemia. Cancer Genet Cytogenet 1999; 111: 166-68.
- Gardiner AC, Corcoran MM, Oscier DG. Cytogenetic, fluorescence in situ hybridisation, and clinical evaluation of translocations with concomitant deletion at 13q14 in chronic lymphocytic leukaemia. Genes Chrom Cancer 1997; 20: 73-81.
- Vanderberghe E, De Wolf Peeters C, Wlodarska I y col. Chromosome 11q rearrangements in B non Hodgkin’s lymphoma. Br J Haematol 1992, 81: 212-17.
- Buhmann R, Kurzeder C, Rehklau J y col. CD40L stimulation enhances the ability of conventional metaphase cytogenetics to detect chromosome aberrations in B-cell chronic lymphocytic leukaemia cells. Br J Haematol 2002; 118: 968-75.
- Kay NE, Suen R, Ranheim E y col. Confirmation of Rb gene defects in B-CLL clones and evidence for variable predominance of the Rb defective cells within the CLL clone. Br J Haematol 1993, 84: 257-64.
- Michaux L, Mecucci C, Stul M y col. BCL3 rearrangement and t(14;18)(32;q21) in lymphoproliferative disorders. Genes Chrom Cancer 1996, 15: 38-47.
- Cuneo A, Roberti MG, Bigoni R y col. Four novel non-random chromosome rearrangements in B-cell lymphocytic leukemia: 6p24-25 and 12p12-13 translocations, 4q21 anomalies and monosomy 21. Brit J Haematol 2000; 108: 559-64.
- Michaux L, Dierlamm J, Wlodarska I y col. t(14;19)/BCL3 rearrangements in lymphoproliferative disorders: a review of 23 cases. Cancer Genet Cytogenet 1997, 94: 36-43.
©
Está
expresamente prohibida la redistribución y la redifusión de todo o parte de los
contenidos de la Sociedad Iberoamericana de Información Científica (SIIC) S.A. sin
previo y expreso consentimiento de SIIC



