EL PAPEL TROMBOGENICO DE UN SISTEMA FIBRINOLITICO ANORMAL
(especial para SIIC © Derechos reservados)
Coautores
Lisa N. Boggio, MD.* Chadi Nabhan, MD*
Division of Hematology and Oncology, Department of Medicine, Northwestern University Feinberg School of Medicine and Robert H. Lurie Comprehensive Cancer Center of Northwestern University, Chicago, IL*
Recepción del artículo: 6 de julio, 2004
Aprobación: 0 de , 0000
 Conclusión breve
El sistema fibrinolítico se encuentra expuesto a numerosos factores que pueden alterar el equilibrio que mantienen la plasmina, sus precursores, los activadores e inhibidores del sistema. Se plantean los conocimientos actuales y el objetivo de las investigaciones en el futuro.
Resumen
Cuando hay un desequilibrio en el sistema fibrinolítico, ya sea adquirido o hereditario, se encuentra aumentado el riesgo de sangrado o de trombosis. Este artículo describe las características de la fibrinólisis anormal que llevan a la trombosis. Los trastornos hereditarios son infrecuentes y pueden afectar la síntesis de plasminógeno, sus activadores y sus inhibidores. Estas anormalidades en la síntesis de plasminógeno y el polimorfismo en el inhibidor de los activadores del plasminógeno tipo 1 (IAP-1) pueden desembocar en eventos tromboembólicos. Los trastornos adquiridos se encuentran en una variedad de enfermedades, principalmente diabetes, cáncer y enfermedades inflamatorias. Los medicamentos también pueden elevar el riesgo de trombosis a través de un aumento en la producción de los inhibidores del plasminógeno o una disminución en la producción de la proteína S. Factores trombofílicos adicionales como deficiencias de la proteína C, de la proteína S y de antitrombina; mutaciones del factor V y la protrombina, y la producción excesiva de factores de la coagulación como los factores VII y II, deben considerarse parte del desarrollo de las condiciones tromboembólicas. El manejo continúa siendo problemático al no haber terapias a largo plazo que restituyan el equilibrio del sistema fibrinolítico.
Palabras clave
Plasminógeno, fibrinólisis, trombosis, cáncer, estados de hipercoagulabilidad
Clasificación en siicsalud
Conclusión breve
El sistema fibrinolítico se encuentra expuesto a numerosos factores que pueden alterar el equilibrio que mantienen la plasmina, sus precursores, los activadores e inhibidores del sistema. Se plantean los conocimientos actuales y el objetivo de las investigaciones en el futuro.
Resumen
Cuando hay un desequilibrio en el sistema fibrinolítico, ya sea adquirido o hereditario, se encuentra aumentado el riesgo de sangrado o de trombosis. Este artículo describe las características de la fibrinólisis anormal que llevan a la trombosis. Los trastornos hereditarios son infrecuentes y pueden afectar la síntesis de plasminógeno, sus activadores y sus inhibidores. Estas anormalidades en la síntesis de plasminógeno y el polimorfismo en el inhibidor de los activadores del plasminógeno tipo 1 (IAP-1) pueden desembocar en eventos tromboembólicos. Los trastornos adquiridos se encuentran en una variedad de enfermedades, principalmente diabetes, cáncer y enfermedades inflamatorias. Los medicamentos también pueden elevar el riesgo de trombosis a través de un aumento en la producción de los inhibidores del plasminógeno o una disminución en la producción de la proteína S. Factores trombofílicos adicionales como deficiencias de la proteína C, de la proteína S y de antitrombina; mutaciones del factor V y la protrombina, y la producción excesiva de factores de la coagulación como los factores VII y II, deben considerarse parte del desarrollo de las condiciones tromboembólicas. El manejo continúa siendo problemático al no haber terapias a largo plazo que restituyan el equilibrio del sistema fibrinolítico.
Palabras clave
Plasminógeno, fibrinólisis, trombosis, cáncer, estados de hipercoagulabilidad
Clasificación en siicsalud
Artículos originales> Expertos del Mundo>
página www.siicsalud.com/des/expertos.php/69188
Especialidades
 Principal: Hematología,
Principal: Hematología,
Relacionadas: Bioquímica, Diagnóstico por Laboratorio, Medicina Interna,
Enviar correspondencia a:
Hau C. Kwaan, MD. Division of Hematology and Oncology, Department of Medicine, Northwestern University Feinberg School of Medicine, 333 East Huron Street, Chicago, IL, EE.UU. Kwaan, Hau C
THE THROMBOGENIC ROLE OF AN ABNORMAL FIBRINOLYTIC SYSTEM
Abstract
When there is an imbalance of the fibrinolytic system, either acquired or hereditary, the risk of bleeding or thrombosis increases. This article describes the characteristics of abnormal fibrinolysis that leads to thrombosis. Hereditary disorders are uncommon and can affect the synthesis of plasminogen, its activators, and its inhibitors. These abnormalities of plasminogen synthesis and polymorphisms in plasminogen activator inhibitor 1 (PAI-1) can lead to thromboembolic events. Acquired disorders are found in a variety of diseases, especially in diabetes, cancer, and inflammatory states. Medications can also increase the risk of thrombosis through increased production of plasminogen inhibitors or decreased production of protein S. Additional thrombophilic factors such as deficiencies of protein C, protein S, and antithrombin; mutations of factor V and prothrombin; and excessive production of clotting factors such as factor VIII and II must be considered in the development of thromboembolic conditions. Management continues to be problematic with no long-acting therapy to counteract an imbalance of the fibrinolytic system.
Key words
Plasminogen, fibrinolysis, thrombosis, cancer, hypercoagulable states
EL PAPEL TROMBOGENICO DE UN SISTEMA FIBRINOLITICO ANORMAL
(especial para SIIC © Derechos reservados)
Artículo completo
Introducción
Una función primaria fisiológica del sistema fibrinolítico es la de mantener el flujo sanguíneo circulante. La insuficiencia en esta función podría resultar en un aumento en el riesgo de trombosis. Por otro lado, un exceso de actividad fibrinolítica podría resultar en una diátesis hemorrágica. Esto fue descubierto tempranamente, cuando Mole estudiaba la fibrinólisis post mortem.1 Mole primero observó que un hombre que había muerto acuchillado en el corazón, no presentaba coágulos post mortem debido a la presencia de la fibrinolisina cadavérica. Posteriormente observó que la fibrinólisis no aparecía en pacientes que morían por infecciones y caquexia. Desde los artículos iniciales, se observó que cierto número de pacientes con déficit heredado del sistema fibrinolítico presentaban alto riesgo de trombosis. En este artículo se consideran tanto las condiciones heredadas como las adquiridas.
El sistema fibrinolítico
Aunque la lisis de fibrina es una de las funciones principales del sistema fibrinolítico, sus componentes se encuentran involucrados es varios procesos biológicos adicionales.2-4 Por eso, resulta más preciso denominarlo sistema plasminógeno-plasmina. Los componentes de este sistema se muestran en la figura 1. La enzima proteolítica plasmina se deriva de la activación de su precursor, el plasminógeno, a través de varios activadores. En el hombre, hay dos activadores del plasminógeno (AP), el AP tipo uroquinasa (APu) y el AP tipo tisular (APt). Los receptores respectivos para el plasminógeno, APu y APt se encuentran presentes en las superficies celulares, facilitando así el ensamblaje del sistema. Sus inhibidores respectivos modulan las actividades proteolíticas de la plasmina y de los AP. Los inhibidores de la plasmina incluyen la alfa 2-antiplasmina y la alfa 2- macroglobulina. Los inhibidores de la AP (IAP) incluyen el tipo 1 (IAP-1), tipo 2 (IAP-2), tipo 3 (IAP-3, idéntico al inhibidor de la proteína C activada) y la nexina de la proteasa. Además, una proteína que se activa durante la formación del trombo, denominada inhibidor de la fibrinólisis activable por trombina (IFAT), también podría inhibir el sistema fibrinolítico.5,6 El IFAT produce una demora en la lisis del coágulo, pero la relevancia clínica de esto es incierta.7
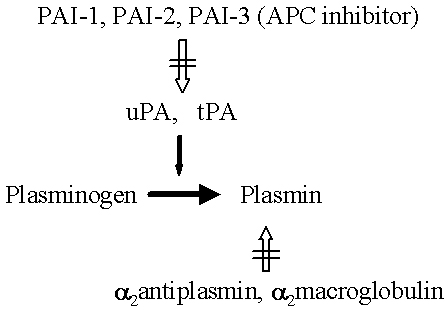
Figura 1. Componentes del sistema plasminógeno-plasmina. La plasmina se forma cuando el plasminógeno se activa por APu o por APt. Esta acción se inhibe por IAP-1, IAP-2 y IAP-3, siendo este último también conocido como inhibidor de la proteína C activada. La plasmina se inhibe por alfa 2-antiplasmina y por alfa 2-macroglobulina.
Una vez producida la brecha en el interior del vaso, la hemostasia normal requiere equilibrio entre la formación de fibrina y su eventual destrucción. Es claro que si la respuesta fibrinolítica tiene lugar demasiado temprano o si hay una excesiva cantidad de proteasa fibrinolítica presente, se producirá una hemorragia. Por otro lado, con el fin de mantener intacto el flujo vascular, el nivel de actividad fibrinolítica debe mantenerse constante a niveles fisiológicos y no puede disminuir. Hay tanto desórdenes congénitos como adquiridos asociados con trastornos de la fibrinólisis en los que el riesgo de trombosis está notablemente aumentado.
Defectos hereditarios
Los defectos hereditarios del sistema fibrinolítico son infrecuentes. Investigaciones recientes que utilizaron ratas knock-out sugieren que una deleción genética de varios componentes de este sistema es compatible con una fisiología funcional y relativamente normal en estos animales.2,8 Esto en parte se debe al concepto “redundante” de que la naturaleza dotó al organismo con más de un sistema de reserva para determinadas funciones. En algunas deficiencias congénitas como el déficit de la alfa 2-antiplasmina, las hemorragias se manifiestan sólo cuando el paciente está estresado, presumiblemente al momento de la liberación de los activadores del plasminógeno en respuesta al estrés. Otros defectos hereditarios se asocian con riesgo aumentado para trombosis o trombofilia. Estos son defectos que pueden involucrar diferentes componentes del sistema, incluyendo el plasminógeno, el activador del plasminógeno y el inhibidor del activador del plasminógeno.
Defectos del plasminógeno
Displasminogenemia tipo I (defecto cuantitativo): El déficit homocigota del plasminógeno se expresa como ausencia de plasminógeno en sangre y tejidos.9 Esto podría resultar en una insuficiencia del organismo para remover los depósitos de fibrina en diversos órganos. Se manifiesta como una conjuntivitis leñosa o seudomembranosa, hidrocefalia (debido a la obstrucción fibrinosa al flujo del LCR), enfermedad obstructiva de las vías respiratorias y trastornos en la cicatrización de las heridas. La terapia de reemplazo con plasminógeno corrige estos defectos, permitiendo la lisis de los depósitos de fibrina. Llamativamente, los niños con el defecto homocigótico no presentan mayor incidencia de eventos tromboembólicos. De igual forma, no hay evidencia convincente que asocie lo heterocigota con el aumento en el riesgo de trombosis. Los heterocigotas afectados presentan disminución de los niveles plasmáticos de plasminógeno. El riesgo de trombosis en la displasminogenemia tipo I es desconocido, así como los reportes presentados en la bibliografía son conflictivos.
Displasminogenemia tipo II (defecto cualitativo): En este trastorno heredado, el nivel de antígenos del plasminógeno es usualmente normal, sin embargo esta proenzima fracasa en su activación. Se hereda como una mutación de varios loci en la molécula de plasminógeno llevando a anormalidades funcionales y a una falta en la activación del plasminógeno. Aunque en varios de los informes originales el defecto en la mutación se detectó cuando el probando se manifestó con eventos tromboembólicos, la mayoría de los miembros afectados en una familia fueron asintomáticos. De esta forma, si la deficiencia congénita del plasminógeno tipo I o II se asocia o no con aumento en la incidencia de trombosis, es algo que aún no pudo comprobarse.
Defectos en el activador del plasminógeno
Hay numerosos artículos previos de familias que presentan trastornos en la liberación de la actividad fibrinolítica luego del esfuerzo o de la oclusión venosa. Se vio que algunos miembros de estas familias presentaban propensión a la trombosis. En lo que respecta a nuestros conocimientos, no se informaron deficiencias congénitas de APu o APt.
Defectos en el inhibidor del activador del plasminógeno
Se observaron tres variaciones polimórficas en el gen humano IAP-1, donde alelos específicos se asociaron con cambios en los niveles de IPA-1 plasmático.10 El primero es un polimorfismo de la longitud del fragmento de restricción Hind III, el segundo es un polimorfismo por repetición de dinucleótidos (C-A)n y el tercero es un polimorfismo por inserción-deleción de nucleótido único (4G/5G). En estudios que evalúan la asociación del polimorfismo del IAP-1 (4G/4G) con la trombosis se publicaron resultados discrepantes. Un metaanálisis indicó que el polimorfismo 4G se asoció con un aumento 1.3 veces mayor en eventos trombóticos coronarios11 a pesar de que no hubo correlación con la trombosis venosa.12
Defectos adquiridos
Diabetes mellitus
Entre pacientes diabéticos la incidencia de enfermedad arterial coronaria, accidentes cerebrovasculares y enfermedad arterial periférica es de 2 a 4 veces mayor que la que se observa en la población no diabética,13,14 transformándola en la causa principal de muerte en diabetes.15 Las anormalidades en la función plaquetaria, los factores de la coagulación y las actividades fibrinolíticas contribuyen globalmente a la patogénesis de la lesión vascular, la aterosclerosis y la trombosis en la diabetes. En especial, la IAP-1 es señalada cada vez más como un factor preponderante de riesgo en la enfermedad vascular.16 Inhibe la apoptosis17 y junto con la APu facilita la migración celular.3,4,18 Cuando son estimuladas por citoquinas, incluyendo IL-1,TNF-alfa, e insulina, muchas células normales, incluso las epiteliales y hepáticas, expresan grandes cantidades de IAP-1.19,20 Datos experimentales tanto in vitro como in vivo indican que el IAP-1 es aterogénico, ya que promueve la migración de células del músculo liso e inhibe la apoptosis. La actividad fibrinolítica plasmática, medida por pruebas globales como el tiempo de lisis de la euglobulina, se ve disminuida principalmente por los niveles elevados de IAP-1. Estudios in vitro demostraron que la insulina, las moléculas tipo proinsulina, la glucosa y las VLDL estimulan la producción de IAP-1.21-23 Los niveles plasmáticos de IAP-1 se correlacionaron fuertemente con la resistencia a la insulina y los valores plasmáticos de insulina. En un estudio multicéntrico que involucró 1 551 individuos se halló correlación estadísticamente significativa entre IAP-1 y los niveles de fibrinógeno y los valores de insulina y sus precursores.24
Variaciones circadianas25
El nivel plasmático de IAP-1 presenta una variación diurna en individuos normales, con valores más elevados en las últimas horas de la mañana. Esto se correlaciona bien con la mayor incidencia de infarto agudo de miocardio,26 accidente vascular27 y muerte súbita cardíaca28 en el mismo período del día. Esta variación circadiana se ve, sin embargo, alterada en los pacientes diabéticos, en los que no presenta aumentos en las horas matutinas25 y sí, en cambio, mayor frecuencia de infartos de miocardio durante las horas de la tarde.29,30
Resistencia a la trombólisis
Se señaló mayor resistencia a la trombólisis en pacientes diabéticos en varios ensayos clínicos sobre terapia trombolítica para el infarto agudo de miocardio y la oclusión arterial periférica.31-33 Las tasas de reperfusión incompleta y de reoclusión fueron mayores en los pacientes diabéticos. Se cree que el aumento de la IAP-1 y la mayor agregación plaquetaria en la diabetes son factores causales en la resistencia a la trombólisis. Sin embargo, debido a que numerosos factores participan de la terapia trombolítica, el papel de los factores trombofílicos permanece poco claro. Igualmente, la evolución de un infarto agudo de miocardio en pacientes diabéticos, tanto a corto como a largo plazo, es peor que en los individuos no diabéticos.
Efecto de las medidas de control metabólico en las anormalidades hemostáticas y fibrinolíticas
No hay ensayos clínicos prospectivos diseñados para disminuir los valores de IAP-1 en diabetes y en síndromes resistentes a la insulina.
Cáncer
Desde las agudas observaciones de Trousseau sobre “phlegmasia alba dolens” en pacientes con “tumores cancerígenos internos”,34 los cambios en los factores hemostáticos de la sangre circulante de los pacientes con cáncer han sido bien demostrados. Estos cambios incluyen niveles anormalmente aumentados de factores de la coagulación y alteraciones de los componentes fibrinolíticos.35-41 En el sistema fibrinolítico se observaron cambios tanto en la prueba global del tiempo de lisis de la euglobulina como en los componentes específicos de los activadores del plasminógeno, el plasminógeno, el IAP-1 y la alfa 2-antiplasmina, al enfrentarse con una actividad fibrinolítica deficitaria. Es de interés comentar que un aumento de los niveles de IAP-1, tanto en sangre como en tejidos tumorales, fue un indicador de mal pronóstico en cánceres de mama, próstata y pulmón.3,42,43 En términos de riesgo trombogénico, el IAP-1 tiene doble efecto. Por un lado dificulta la activación del plasminógeno, aumentando de esa forma el riesgo de tromboembolismo; por otro lado inhibe la apoptosis.17 Recientemente se observó que las células apoptóticas generan trombina,44 este acto de IAP-1 indirectamente disminuye la trombogenicidad de las células tumorales.
Las células tumorales frecuentemente contienen inhibidores de la fibrinólisis. Esto se observó primero en el carcinoma hepatocelular.45 Más recientemente se vio que el IAP-1 era uno de los inhibidores en varios tipos tumorales, incluidos los cánceres de mama, próstata, colon y el carcinoma de células escamosas de la piel.3 No está comprobado si las anormalidades celulares contribuyen o no al riesgo trombótico en los pacientes con cáncer. Sin embargo, la actividad fibrinolítica aumentada contribuye a las complicaciones hemorrágicas en la leucemia promielocítica aguda.46
Clínicamente, el tromboembolismo es la segunda causa más frecuente de muerte en los pacientes con cáncer.47 Entre la multitud de factores trombogénicos, juegan un papel primordial la expresión del factor tisular y los procoagulantes cancerígenos de las células tumorales. En más de tres cuartos de los pacientes estudiados se hallaron evidencias de coagulación activa, como indica la presencia de niveles de fibrinopéptido A.48 Como resultado, se requiere una adecuada respuesta fibrinolítica para prevenir la trombosis. De esta forma, los niveles de inhibidores de la fibrinólisis que se encontraron en paciente con cáncer podrían ser esenciales para determinar la aparición de esta complicación. En el cáncer se presentan trastornos del equilibrio fisiológico entre los activadores del plasminógeno y sus inhibidores, tanto a nivel celular como en sangre circulante.3 El desequilibrio presente favorece la trombosis. Tanto el activador del plasminógeno tipo uroquinasa (APu) como el inhibidor del activador de plasminógeno tipo I (IAP-1) se expresan en forma importante en varios tumores y son indicadores de mal pronóstico. Las células mieloides normales cambian su producción de activadores del plasminógeno de APt a APu cuando maduran.49 En la leucemia mielocítica aguda, tanto el APu como el APt pueden hallarse en las células leucémicas, pero una expresión elevada del APt indica mal pronóstico. Los cambios fibrinolíticos fueron estudiados más ampliamente en la leucemia promielocítica aguda. La sangre de estos pacientes presenta evidencias de coagulación intravascular diseminada (descenso de fibrinógeno y antitrombina, aumento de los productos de la degradación de la fibrina-dímero-D, fibrinopéptido A, fragmento 1+2 de protrombina, complejo trombina-antitrombina) y fibrinólisis (aumento de APt y APu),50 y descenso de alfa 2-antiplasmina.51 El descenso de alfa 2-antiplasmina se cree que se debe a su inhibición por las elastasas leucocitarias.52 Se halló recientemente que el IAP-1 se encontraba elevado en la leucemia promielocítica aguda, agregando así otro factor a la compleja coagulopatía que se observa en esta enfermedad.53 Así, hay hipercoagulabilidad que lleva a coagulacion intravascular diseminada y a aumento de la fibrinólisis, lo que resulta en complicaciones hemorrágicas. El aumento de IAP-1 tiene lugar al momento del diagnóstico de la enfermedad y es probablemente secundario a la respuesta del organismo a las citoquinas inflamatorias, y podría desempeñar un papel en la supresión de la fibrinólisis, al contribuir a la trombofilia en una etapa posterior de la enfermedad.
Tanto el APu como el APt tienen efectos adicionales sobre el sistema fibrinolítico. Estos activadores del plasminógeno activan varias citoquinas latentes, incluyendo bFGF y TGF-beta.54 Estas citoquinas aumentan por etapas la producción de IAP-1. Un nivel elevado de IAP-1 en sangre incrementa el riesgo de trombosis. En los linfomas, especialmente en el subtipo escleronodular de la enfermedad de Hodgkin, las células tumorales producen grandes volúmenes de TGF-beta, llevando a niveles elevados de IAP-1, que favorecen así la trombosis. Las citoquinas inflamatorias, como el FNT-alfa y la IL-1-beta también aumentan el IAP-1 y el TF en las células endoteliales, agregando otro factor al riesgo trombótico. La actividad fibrinolítica disminuida también podría ser resultado del uso de la L-asparaginasa en el tratamiento del cáncer linfoideo.
La L-asparaginasa utilizada en el tratamiento del cáncer linfocítico, incluida la leucemia linfoblástica aguda y algunos casos de linfoma no Hodgkin, se vio implicada como factor de riesgo en la enfermedad tromboembólica. Se piensa que se produciría a través de la inhibición de la síntesis proteica, disminuyendo así los niveles plasmáticos de plasminógeno, antitrombina y varios factores de la coagulación.55,56 Kucuk y col.57 describieron un caso de leucemia linfoblástica aguda en el cual una mujer de 18 años presentó ACV y trombosis de la vena subclavia asociado al catéter, luego de haber recibido L-asparaginasa. Se observó al momento de los eventos tromboembólicos, que presentaba bajos niveles de plasminógeno y antitrombina. La terapia trombolítica para la trombosis venosa con un activador del plasminógeno no fue exitosa debido a los bajos valores del plasminógeno y hasta su corrección por infusión de plasma fresco congelado.
Inflamación aguda y crónica
El riesgo de enfermedades tromboembólicas está aumentado tanto en los trastornos inflamatorios agudos como crónicos. La incidencia de eventos tromboembólicos en la enfermedad intestinal inflamatoria crónica ha sido descrita con una amplitud del 0.4% al 39%.58 Estos eventos son característicamente recurrentes, con un margen del 10% al 13%.59 A pesar de que los factores trombogénicos principales son el aumento de los factores de la coagulación (factores V, VIII, XIII) con un descenso en los inhibidores de la coagulación (proteínas C y S, antitrombina), el sistema fibrinolítico también se ve afectado. Esto es debido a un aumento en el IAP-1 y la antiplasmina,60 mediante un aumento en la expresión de IAP-1 a través de las citoquinas inflamatorias (TNF alfa e IL-1b).
Efectos hormonales
La terapia de reemplazo hormonal con dosis fisiológicas de estrógenos se asocia con descenso de los eventos cardiovasculares y ACV.61 Hay un aumento de APt junto con la disminución de los valores de IAP-1 que resulta en un aumento global de la actividad fibrinolítica.62 Por otro lado, está claro que las dosis farmacológicas de estrógeno63 y los modificadores de los receptores selectivos de estrógenos (SERMS) utilizados en pacientes con cáncer se asocian con aumento en el riesgo de eventos tromboembólicos,64,65 principalmente cuando se utilizan combinados con quimioterapia.66 El papel del sistema fibrinolítico en esto es poco claro. Los estrógenos ejercen efectos sobre la pared vascular, pudiendo afectar de esta forma en dosis fisiológicas la liberación de los activadores del plasminógeno de las células endoteliales. Es interesante observar que con el uso de anticonceptivos orales los cambios en el nivel de varios componentes del sistema fibrinolítico, incluidos el plasminógeno, el activador del plasminógeno tisular, el inhibidor del activador del plasminógeno tipo 1 y los complejos plasmina-antiplasmina indicaron que está aumentada la actividad fibrinolítica global.65,67-70 Llamativamente, los valores del inhibidor de la fibrinólisis activable por trombina (IFAT) están aumentados en mujeres que toman anticonceptivos que contienen desorgestrel,71 lo cual sugierie que cuando se produce la coagulación, el IFAT desempeña un papel al inhibir la fibrinólisis. Esto lo confirman los estudios que demuestran incidencia aumentada de trombosis en las mujeres con el gen mutante del factor V de Leiden, que toman compuestos que contienen desorgestrel.72
Manejo
Cuando se sospecha que un evento trombótico está vinculado con una alteración del sistema fibrinolítico, el manejo es generalmente sintomático. Ya que múltiples factores de riesgo tromboembólico podrían presentarse en un paciente determinado, es prudente determinar cualquier causa reversible, así como instituir una anticoagulación apropiada.
Conclusión
El sistema fibrinolítico desempeña un papel fundamental en la regulación de la hemostasis y en la prevención de la trombosis. Hasta el momento no hay drogas disponibles que puedan aumentar en forma efectiva la actividad fibrinolítica del plasma por tiempo prolongado como para prevenir eventos trombóticos. A pesar de la habilidad de los agentes vasoactivos como el ácido nicotínico y la metformina para liberar el activador del plasminógeno de las paredes vasculares, este efecto terapéutico no fue evaluado adecuadamente. Los activadores del plasminógeno son todos de acción breve y están indicados sólo para la trombólisis y no para profilaxis. Los estudios futuros se orientan hacia el hallazgo de agentes que puedan tanto aumentar la liberación de los activadores del plasminógeno como inhibir la actividad del PAI-1.
Los autores no manifiestan conflictos.
Bibliografía del artículo
- Mole R. Fibrinolysin and the fluidity of blood post mortem. J Path & Bact 1948;60:413-427.
- Kwaan H, McFadzean A, Cook J. Plasma fibrinolytic activity in cirrhosis of the liver. Lancet 1952; i:132-156.
- Kwaan H, McFadzean A. The inhibition of clot lysis by corticotrophin. Lancet 1956;i:136-137.
- Kwaan H, Lo R, McFadzean A, et al. On plasma fibrinolytic activity in cryptogenic splenomegaly. Clin Sci 1959;18:251-261.
- Kwaan H, Lo R, McFadzean A. Antifibrinolytic activity in primary carcinoma of the liver. Clin Sci 1959;18:251-261.
- Bachmann F. Plasminogen-plasmin enzyme system. In: Coleman R, Hirsh J, Marder V, et al., editors. Hemostasis and Thrombosis, Basic Principles and Clinical Practice. Philadelphia: Lippincott Williams & Wilkins; 2001. p. 275-320.
- Kwaan H. The plasminogen-plasmin system in malignancy. Cancer Met Rev 1992;11:291-311.
- Kwaan H. The biologic role of components of the plasminogen-plasmin system. Prog Cardiovascular Dis 1992;34:309-316.
- Hendriks D, Scharpe S, Sande Mv, et al. Characterization of carboxypeptidase in human serum distinct from carboxypeptidase N. J Clin Chem Clin Biochem 1989;27:277-285.
- Juhan-Vague I, JF JR, Grimaux M, et al. Thrombin-activatable fibrinolysis inhibitor antigen levels in cardiovascular risk factors. Arterioscler Thromb Vasc Biol 2000;20:2156-2161.
- Nagashima M, Yin Z, Zhao L, et al. Thrombin-activatable fibrinolysis inhibitor (TAFI) deficient mice. J Clin Invest 2002;109:101-10.
- Nagashima M, Yin Z, Zhao L, et al. Thrombin-activatable fibrinolysis inhibitor (TAFI) deficiency is compatible with murine life. J Clin Invest 2002;109:101-110.
- Raum D, Marcus D, Alper C, et al. Synthesis of human plasminogen by the liver. Science 1980;208:1036-1037.
- Collen D, DeMaeyer L. Molecular biology of human plasminogen: Physicochemical properties and microheterogeneity. Thromb Diath Haemorrh 1975;34:396-402.
- Ji W-R, Castellino F, Y YC, et al. Charactization of kringle domains of angiostatin as antagonists of endothelial cell migration, an important process in angiogenesis. FASEB J 1998;12:1731-1738.
- Mingers A, Heimburger N, Zeitler P, et al. Homozygous type 1 plasminogen deficiency. Semin Thromb Hemost 1997;23:259-269.
- Shigekiyo T, Uno Y, Tomonari A, et al. Type 1 congenital plasminogen deficiency is not a risk factor for thrombosis. Thromb Haemost 1992; 67:189-192.
- Tait R, Walker I, Conkie J, et al. Isolated familial plasminogen deficiency may not be a risk factor for thrombosis. Thromb Haemost 1996;76:1004-1008.
- Stolz E, Kemkes-Matthes B, Potzsch B, et al. Screening for thrombophilic risk factors among 25 German patients with cerebral venous thrombosis. Acta Neurologica Scandinavica 2000;102:31-6.
- Iacoviello L, Burzotta F, DiCastelnuovo A, et al. The 4G/5G polymorphism of PAI-1 promotor gene and the risk of myocardial infarction: a meta-analysis. Thromb Haemost 1998;80:1029-1030.
- Mayata T, Iwananga S, Sakata Y, et al. Plasminogen Tochigi: Inactive plasmin resulting from replacement of alanine-600 by threonine in the active site. Proc Natl Acad Sci 1982;79:6132-6136.
- Tsutsumi S, Saito T, Sakata T, et al. Genetic diagnosis of dysplasminogenia: Detection of an Ala601-Thr mutation in 118 out of 125 families and identification of a new Asp676-Asn mutation. Thromb Haemost 1996;76:135-138.
- Mima N, Azuma H, Shigekiyo T, et al. A novel missense in two families with congenital plasminogen deficiency-identification of an Ala (675) to Thr (675) substitution. Thromb Haemost 1996;75:96-100.
- Kohler H, Grant P. Plasminogen activator type 1 and coronary artery disease. N Eng J Med 2000;342:1792-1801.
- Dawson S, Hamsten A, Wilman B, et al. Genetic variation at the plasminogen activator inhibitor-1 locus is associated with altered levels of plasma plasminogen activator inhibitor-1 activity. Arterioscl Thromb 1991;11:183-190.
- Eriksson P, Kallin B, Hooft FVt, et al. Allele-specific increase in basal transcription of the plasminogen-activator gene is associated with myocardial infarction. Proc Natl Acad Sci 1995;92:1851-1855.
- Ye S, Green F, Scarabin P, et al. The 4G/5G genetic polymorphism in the promotor of the plasminogen activator inhibitor-1 (PAI-1) gene is associated with differences in plasma PAI-1 activity but not with the risk of myocardial infarction in the ECTIM study. Thromb Haemost 1995;74:837-841.
- Ridker P, Hennekens C, Lindpainter K, et al. Arterial and venous thrombosis is not associated with the 4G/5G polymorphism in the promoter of the plasminogen activator inhibitor gene in a large cohort of U.S. men. Circulation 1997;95:59-62.
- Zoller B, Frutos PGd, Dahlback B. A common 4G allele in the promotor of the plasminogen activator inhibitor-1 (PAI-1) gene is a risk factor for pulmonary embolism and arterial thrombosis in hereditary protein S deficiency. Thromb Haemost 1998;79:802-807.
- Ichinose A, Espiling E, Takamatsu J, et al. Two types of abnormal genes for plasminogen in families with a predisposition for thrombosis. Proc Natl Acad Sci 1991;88:115-119.
- Folsom A, Cushman M, Heckbert S, et al. Prospective study of fibrinolytic markers and venous thromboembolism. J Clin Epi 2003;56:598-603.
- Morrish N, Stevens L, Head J, et al. A prospective study of mortality among middle-aged diabetic patients (the London cohort of the WHO multinational study of vascular disease in diabetics). I. Causes and death rates. Diabetologia 1990;33:538-541.
- Stamler J, Vaccaro O, Neaton J. Diabetes, other risk factors and 12-year cardiovascular mortality of men screened in the multiple risk factor intervention trial. Diabetes Care 1993;16:434-444.
- Anonymous. Consensus Statement. Role of cardiovascular risk factors in the prevention and treatment of macrovascular disease in diabetes. Diabetes Care 1989;12:573-579.
- Vaughan D. Plasminogen activator inhibitor-1: a common denominator in cardiovascular disease. J Invest Med 1998;46:370-376.
- Kwaan H, Wang J, Svoboda K, et al. Plasminogen activator 1 may promote tumor growth through the inhibition of apoptosis. Br J Cancer Met Rev 2000;82:1702-1708.
- Collen D. The plasminogen (fibrinolytic) system. Thromb. Haemost 1999;82:259-270.
- Alessi M, Juhan-Vague I, Koiostra T, et al. Insulin stimulates the synthesis of plasminogen activator inhibitor 1 by the human hepatocellular cell line HepG2. Thromb Haemost 1988;60:491-494.
- Juhan-Vague I, Alessi M, Vague P. Increases plasma plasminogen activator inhibitor l levels. A possible link between insulin resistance and atherothrombosis. Diabetologia 1991;34:457-462.
- Li X, Grenett H, Benza R, et al. Genotype-specific transcriptional regulation of PAI-1 expression by hypertriglyceridemic VLDL and Lp (a) in cultured human endothelial cells. Aterioscl Thromb Vasc Biol 1997;17:3215-3223.
- Nort T, Schneider D, Sobel B. Augmentation of the synthesis of plasminogen activator inhibitor type-1 by precursors of insulin. A potential risk factor for cardiovascular disease. Circulation 1994;89:321-330.
- Sironi L, Mussoni L, Prati L, et al. Plasminogen activator inhibitor type-1 synthesis and mRNA expression in HepG2 cells are regulated by VLDL. Arterioscl Thromb Vas Biol 1996;16:89-96.
- Chen Y, Su M, Walia R, et al. Sp1 sites mediate activation of the plasminogen-activator inhibitor-1 promotor by glucose in smooth muscle cells. J Biol Chem 1998;273:8225-8231.
- Calles-Escandon L, Mirza S, Sobel B, et al. Induction of hyperinsulinemia combined with hyperglycemia and hypertrygliceridemia increase plasminogen activator inhibitor 1 in blood of normal human subjects. Diabetes Care 1998;47:292-293.
- Festa A, D'Augistino R, Mykkanen L, et al. Relative contribution of insulin and its precursors to fibrinogen and PAI-1 in a large population with different states of glucose tolerance: the Insulin Resistance Atherosclerosis Study (IRAS). Arterioscl Thromb Vas Biol 1999;19:562-568.
- Sobel B, Woodcock-Mitchell J, Schneider D, et al. Increase plasminogen activator type 1 in coronary artery atherectomy specimens from type 2 diabetic compared with non diabetic patients: a potential factor predisposing to thrombosis and its persistence. Circulation 1998;97:2213-2221.
- Aronson D, Weinrauch L, D'Elia J, et al. Circadian patterns of heart rate viability, fibrinolytic activity, and hemostatic factors in type-1 diabetes mellitus with autonomic neuropathy. Am J Cardiol 1999;84:449-455.
- Muller J, Ludmer P, Willich S, et al. Circadian variation in the frequency of sudden cardiac death. Circulation 1987;75:131-138.
- Tsementzis S, Gill J, Hitchcock E, et al. Diurnal variation of, and activity during, the onset of stroke. Neurosurgery 1985;17:901-907.
- Muller J, Stone P, Turi Z, et al. Circadian variation in the frequency of onset of acute myocardial infarction. N Engl J Med. 1985;313:1315-1322.
- Fava S, Azzopardi J, Muscat H, et al. Absence of circadian variation in the onset of acute myocardial infarction in diabetic subjects. Br Heart J 1995;74:370-372.
- Tanka T, Fujita M, Fudo T, et al. Modification of the circadian variation of symptom onset of acute myocardial infarction in diabetes mellitus. Coronary Artery Dis 1995;6:241-244.
- Gray R, Yudkin J, Patterson D. Enzymatic evidence of impaired reperfusion in diabetic patients after thrombolytic therapy for acute myocardial infarction: a role for plasminogen activator inhibitor Br Heart J 1993;70:530-536.
- Ouriel K, Shortell C, Azodo M, et al. Acute peripheral arterial occlusion: predictors of success in catheter-directed thrombolytic therapy. Radiology 1994;193:561-566.
- Zuanetti G, Latini R, Maggioni A, et al. Influence of diabetes on mortality in acute myocardial infarction: data from the GISSI-2 study. J Am Coll Cardiol 1993;22:1788-1794.
- Tan K, Janus E, Lam K. Effects of fluvastatin on prothrombotic and fibrinolytic factors in type 2 diabetes mellitus. In J Cardiol 1999;84:934-937.
- Kruszynska Y, Yu J, Olesfky J, et al. Effects of troglitazone on blood concentrations of plasminogen activator inhibitor 1 in patients with type 2 diabetes and in lean and obese normal patients. Diabetes Care 2000;49:633-639.
- Trousseau A. In: Clinique Medicale de Hotel Dieu de Paris. 2nd ed; 1865. p. 654-712.
- Brugarolas A, Mik I, Elias E, et al. Correlation of hyperfibrinogenemia with major thromboembolism in patients with cancer. Surg Gynecol Obstet 1981;136:75-77.
- Davis R, Theologides A, Kennedy B. Comparitive studies of blood coagulation and platelet aggregation in patients with cancer and non-malignant diseases. Ann Int Med 1969;71:67-80.
- DeJong E, Knot E, Piker D, et al. Increased plasminogen activator activity in malignancy. Thromb Haemost 1987;57:140-143.
- Miller S, Sanchez-Avalos J, Stefanski T. Coagulation disorders in cancer. I. Clinical and laboratory studies. Cancer Met Rev 1967;20:1452-1465.
- Paramo R, Fernandez F, Cuesta B, et al. Clotting activity and impairment of fibrinolysis in malignancy. Thromb Res 1989;54:699-707.
- Rennie J, Ogston D. Fibrinolytic activity in malignant disease. J Clin Path 1975;28:872-874.
- Slichter S, Harker L. Hemostasis in malignancy. Ann NY Acad Sci 1974;230:252-261.
- Ozyikan O, Baltali E, Ozdemir O, et al. Haemostatic changes: Plasma levels of alpha 2 antiplasmin-plasmin complex and thrombin-antithrombin complex in female breast cancer. Tumori 1998;84:364-367.
- Taguchi O, Gabazza E, Yoshida M, et al. High plasma level of plasmin-alpha 2-plasmin inhibitor complex is a predictor of poor prognosis with lung cancer. Clin Chim Acta 1996;244:69-81.
- Wang J, Weiss I, Svoboda K, et al. Thrombogenic role of cells undergoing apoptosis. Br J Haematol 2001;115:382-391.
- Kwaan HC, Wang J, Boggio LN. Abnormalities in hemostasis in acute promyelocytic leukemia. Hematol Oncol 2002;20(1):33-41.
- Kwaan HC. Hypercoagulability and cancer. In: Smama M, Seghatchian M, Hecker S, editors. Hypercoagulable states: Fundamental aspects, acquired disorders, and congenital thrombophilia. Boca Raton: CRC Press; 1996. p. 317-334.
- Rickles FR, Levine M, Edwards RL. Hemostatic alterations in cancer patients. Cancer Metastasis Rev 1992;11(3-4):237-48.
- Wilson EL, Francis GE. Differentiation-linked secretion of urokinase and tissue plasminogen activator by normal human hemopoietic cells. J Exp Med 1987;165(6):1609-23.
- Bennett B, Booth N, Croll A, et al. The bleeding disorder in acute promyelocytic leukemia: Fibrinolysis due to u-PA rather than defibrination. Br J Haematol 1989;71:511-517.
- Avvisati G, Ten Cate J, Sturk A, et al. Acquired alpha-2-antiplasmin deficiency in acute promyelocytic leukemia. Br J Haematol 1988;70:43-48.
- Brower N, Harpel P. Proteolytic cleavage and inactivation alpha-2-plasmin inhibitor and C1 inhibitor by human polymorphonuclear leukocyte elastase. J Biol Chem 1982;257:9849-9854.
- Falanga A, Iacoviello L, Evangelista V, et al. Loss of blast cell procoagulant activity and improvement of hemostatic variables in patients with acute promyelocytic leukemia administered all-trans-retinoic acid. Blood 1995;86(3):1072-81.
- Lyons RM, Keski-Oja J, Moses HL. Proteolytic activation of latent transforming growth factor-beta from fibroblast-conditioned medium. J Cell Biol 1988;106(5):1659-65.
- Priest JR, Ramsay NK, Bennett AJ, et al. The effect of L-asparaginase on antithrombin, plasminogen, and plasma coagulation during therapy for acute lymphoblastic leukemia. J Pediatr 1982;100(6):990-5.
- Sills RH, Nelson DA, Stockman JA, 3rd. L-Asparaginase-induced coagulopathy during therapy of acute lymphocytic leukemia. Med Pediatr Oncol 1978;4(4):311-3.
- Kucuk O, Kwaan HC, Gunnar W, et al. Thromboembolic complications associated with L-asparaginase therapy. Etiologic role of low antithrombin III and plasminogen levels and therapeutic correction by fresh frozen plasma. Cancer 1985;55(4):702-6.
- van Bodegraven AA. Haemostasis in inflammatory bowel diseases: clinical relevance. Scand J Gastroenterol Suppl 2003(239):51-62.
- Jackson LM, O'Gorman PJ, O'Connell J, et al. Thrombosis in inflammatory bowel disease: clinical setting, procoagulant profile and factor V Leiden. Qjm 1997;90(3):183-8.
- Van Bodegraven AA, Tuynman HA, Schoorl M, et al. Fibrinolytic split products, fibrinolysis, and factor XIII activity in inflammatory bowel disease. Scand J Gastroenterol 1995;30(6):580-5.
- Idell S. Coagulation, fibrinolysis, and fibrin deposition in acute lung injury. Crit Care Med 2003;31(4 Suppl):S213-20.
- Wild RA, Reis SE. Estrogens, progestins, selective estrogen receptor modulators, and the arterial tree. Am J Obstet Gynecol 2001;184(5):1031-9.
- Gilabert J, Estelles A, Cano A, et al. The effect of estrogen replacement therapy with or without progestogen on the fibrinolytic system and coagulation inhibitors in postmenopausal status. Am J Obstet Gynecol 1995;173(6):1849-54.
- Inman WH, Vessey MP. Investigation of deaths from pulmonary, coronary, and cerebral thrombosis and embolism in women of child-bearing age. Br Med J 1968;2(599):193-9.
- Fisher B, Costantino JP, Wickerham DL, et al. Tamoxifen for prevention of breast cancer: report of the National Surgical Adjuvant Breast and Bowel Project P-1 Study. J Natl Cancer Inst 1998;90(18):1371-88.
- Vandenbroucke JP, Rosing J, Bloemenkamp KW, et al. Oral contraceptives and the risk of venous thrombosis. N Engl J Med 2001;344(20):1527-35.
- Levine MN, Gent M, Hirsh J, et al. The thrombogenic effect of anticancer drug therapy in women with stage II breast cancer. N Engl J Med 1988;318(7):404-7.
- Bonnar J. Coagulation effects of oral contraception. Am J Obstet Gynecol 1987;157(4 Pt 2):1042-8.
- Kluft C, Lansink M. Effect of oral contraceptives on haemostasis variables. Thromb Haemost 1997;78(1):315-26.
- Stubblefield PG. Cardiovascular effects of oral contraceptives: a review. Int J Fertil 1989;34 Suppl:40-9.
- Winkler UH. Blood coagulation and oral contraceptives. A critical review. Contraception 1998;57(3):203-9.
- Meijers JC, Middeldorp S, Tekelenburg W, et al. Increased fibrinolytic activity during use of oral contraceptives is counteracted by an enhanced factor XI-independent down regulation of fibrinolysis: a randomized cross-over study of two low-dose oral contraceptives. Thromb Haemost 2000;84(1):9-14.
- Bloemenkamp KW, Rosendaal FR, Helmerhorst FM, et al. Enhancement by factor V Leiden mutation of risk of deep-vein thrombosis associated with oral contraceptives containing a third-generation progestagen. Lancet 1995;346(8990):1593-6.
- Cesarman G, Rios N, Sanchez-Guerrero J, et al. Antibodies to annexin II-a fibrinolytic receptor - are highly prevalent in antiphospholipid syndrome (APS) and may be related to thrombosis. Arthritis Rheum 2001;44(Suppl):S74.
©
Está
expresamente prohibida la redistribución y la redifusión de todo o parte de los
contenidos de la Sociedad Iberoamericana de Información Científica (SIIC) S.A. sin
previo y expreso consentimiento de SIIC



