MECANISMOS DE ACCION IMPLICADOS EN LOS EFECTOS ANTIDIABETICOS DE LAS TIAZOLIDINEDIONAS
(especial para SIIC © Derechos reservados)
Coautores
Mireia Jové Godino* Anna Planavila**
Licenciada en Farmacia, Universidad de Barcelona*
Licenciada en Biología, Universidad de Barcelona**
Recepción del artículo: 21 de abril, 2004
Aprobación: 2 de septiembre, 2004
 Conclusión breve
El mecanismo de acción responsable de los efectos antidiabéticos de las tiazolidinedionas radica en la aparente paradoja que existe entre la utilización de fármacos antidiabéticos que favorecen la adipogénesis para el tratamiento de la diabetes tipo 2, cuando el factor de riesgo más importante para el desarrollo de esta patología es la obesidad.
Resumen
Las tiazolidinedionas (TZD), también llamadas glitazonas, son una nueva clase de fármacos antidiabéticos que han sido introducidos recientemente para el tratamiento de la diabetes mellitus tipo 2. Las TZD fueron la primera clase de compuestos identificados como ligandos de los receptores activados por proliferadores de peroxisomas γ (peroxisome proliferator-activated receptors, PPAR). La participación de los PPARγ en los efectos farmacológicos de las TZD se basó en estudios que demostraron una excelente correlación entre el efecto hipoglucemiante de estos fármacos y su afinidad por PPARγ. Aunque el músculo es responsable de la utilización de hasta un 80% de la glucosa estimulada por la insulina, se considera que el tejido adiposo es el lugar de acción principal de las TZD. Respecto del mecanismo de acción responsable de los efectos antidiabéticos de las TZD, recientes estudios realizados en animales parecen explicar la aparente paradoja que existe entre la utilización de fármacos antidiabéticos que favorecen la adipogénesis para el tratamiento de la diabetes tipo 2, cuando el factor de riesgo más importante para el desarrollo de esta patología es la obesidad. El objetivo de este artículo es revisar los estudios más relevantes realizados para determinar el mecanismo de acción de las TZD antidiabéticas.
Palabras clave
Tiazolidinedionas, diabetes tipo 2, PPARγ, tejido adiposo, músculo esquelético
Clasificación en siicsalud
Conclusión breve
El mecanismo de acción responsable de los efectos antidiabéticos de las tiazolidinedionas radica en la aparente paradoja que existe entre la utilización de fármacos antidiabéticos que favorecen la adipogénesis para el tratamiento de la diabetes tipo 2, cuando el factor de riesgo más importante para el desarrollo de esta patología es la obesidad.
Resumen
Las tiazolidinedionas (TZD), también llamadas glitazonas, son una nueva clase de fármacos antidiabéticos que han sido introducidos recientemente para el tratamiento de la diabetes mellitus tipo 2. Las TZD fueron la primera clase de compuestos identificados como ligandos de los receptores activados por proliferadores de peroxisomas γ (peroxisome proliferator-activated receptors, PPAR). La participación de los PPARγ en los efectos farmacológicos de las TZD se basó en estudios que demostraron una excelente correlación entre el efecto hipoglucemiante de estos fármacos y su afinidad por PPARγ. Aunque el músculo es responsable de la utilización de hasta un 80% de la glucosa estimulada por la insulina, se considera que el tejido adiposo es el lugar de acción principal de las TZD. Respecto del mecanismo de acción responsable de los efectos antidiabéticos de las TZD, recientes estudios realizados en animales parecen explicar la aparente paradoja que existe entre la utilización de fármacos antidiabéticos que favorecen la adipogénesis para el tratamiento de la diabetes tipo 2, cuando el factor de riesgo más importante para el desarrollo de esta patología es la obesidad. El objetivo de este artículo es revisar los estudios más relevantes realizados para determinar el mecanismo de acción de las TZD antidiabéticas.
Palabras clave
Tiazolidinedionas, diabetes tipo 2, PPARγ, tejido adiposo, músculo esquelético
Clasificación en siicsalud
Artículos originales> Expertos del Mundo>
página www.siicsalud.com/des/expertos.php/68042
Especialidades
 Principal: Endocrinología y Metabolismo,
Principal: Endocrinología y Metabolismo,
Relacionadas: Bioquímica, Farmacología, Inmunología, Medicina Farmacéutica, Nutrición,
Enviar correspondencia a:
Manuel Vázquez Carrera. Unidad de Farmacología. Facultad de Farmacia. Diagonal 643 E-08028, Barcelona, España. Vázquez-Carrera, Manuel
Patrocinio y reconocimiento
Los estudios realizados en el Departamento de Farmacología y Química Terapéutica fueron subvencionados por la Fundació Privada Catalana de Nutrició i Lípids (FPCNL) y por el Ministerio de Ciencia y Tecnología de España (SAF00-0201, SAF 2003-01232 y BFI02-05167). También agradecemos a la Generalitat de Catalunya la subvención 2001SGR00141. Mireia Jové recibió una beca FPI del Ministerio de Ciencia y Tecnología de España. Anna Planavila cuenta con una beca de la División IV de la Universidad de Barcelona.
MECHANISMS OF ACTION INVOLVED IN THE ANTIDIABETIC EFFECTS OF THIAZOLIDINEDIONES
Abstract
Thiazolidinediones (TZD), also called glitazones, are a new class of antidiabetic drugs that have recently been introduced as therapeutic agents for the treatment of type 2 diabetes mellitus. TZD were the first class of compounds to be identified as peroxisome proliferator-activated receptors (PPAR) γ-ligands. The involvement of PPARγ in the pharmacological effects of TZD was supported by studies showing an excellent correlation between the hypoglycemic action of these drugs and their affinity for PPARγ. Despite this evidence, the site of action and the molecular mechanism of TZD remains unclear. Although up to 80% of insulin-stimulated glucose disposal in humans occurs in skeletal muscle, the primary site of action of TZD is thought to be the adipose tissue. Regarding the mechanism of action responsible for the antidiabetic effects of TZD, recent studies performed in animals seem to explain the apparent paradox that lies in using antidiabetic drugs that promote adipogenesis for the treatment of type 2 diabetes, when the major risk factor for the development of this pathology is obesity. The aim of the present article is to review the most relevant studies performed in the search for establishing the mechanism of action of antidiabetic TZD.
Key words
Thiazolidinediones, type 2 diabetes mellitus, PPARγ, adipose tissue, skeletal muscle
MECANISMOS DE ACCION IMPLICADOS EN LOS EFECTOS ANTIDIABETICOS DE LAS TIAZOLIDINEDIONAS
(especial para SIIC © Derechos reservados)
Artículo completo
Introducción
Las tiazolidindionas (TZD) son una nueva clase de fármacos antidiabéticos recientemente introducidos como agentes terapéuticos para el tratamiento de la diabetes mellitus tipo 2.1-3 Dentro de este grupo de antidiabéticos se incluyen fármacos como la troglitazona, la rosiglitazona y la pioglitazona que fueron desarrollados a través de una selección empírica en modelos animales de resistencia a la insulina.4 En estos modelos animales de resistencia a la insulina las TZD, también llamadas glitazonas, disminuyeron los niveles de glucosa y de insulina en plasma y mejoraron algunas anormalidades del metabolismo lipídico. Basados en estos esperanzadores resultados en animales se llevaron a cabo muchos estudios clínicos en pacientes diabéticos tipo 2. En estos pacientes, las TZD disminuyeron los niveles de glucosa y de insulina en suero plasmático, aumentaron la recaptación periférica de glucosa y disminuyeron los niveles de triglicéridos.1 El mecanismo de acción molecular de las TZD permaneció desconocido hasta que diversos estudios demostraron que estos agentes inducían la transcripción de ciertos genes en el tejido adiposo.5,6 En cultivos celulares, el tratamiento con TZD incrementaba la expresión de genes implicados en la diferenciación adipocitaria.5,7 La región promotora de uno de estos genes cuya expresión es incrementada por las TZD, el marcador adipocitario aP2 (adipocyte-specific fatty acid-binding protein), es conocido por su capacidad para unirse a factores de transcripción denominados PPARγ y esto llevó a diversos investigadores a proponer que las TZD ejercían sus efectos por activación de estos receptores nucleares.7-10 Los receptores activados por proliferadores de peroxisomas (peroxisome proliferator-activated receptors [PPAR]) son miembros de una superfamilia de receptores hormonales que actúan como factores de transcripción activados por ligandos y que están relacionados con los receptores hormonales de los retinoides, hormonas esteroideas y tiroideas.
La subfamilia de los PPAR está formada por tres subtipos, PPARα (NR1C1, según el sistema de nomenclatura unificado para la superfamilia de receptores nucleares), PPARδ/β (NR1C2) y PPARγ (NR1C3).11 Los PPARα se expresan principalmente en tejidos con elevado nivel de catabolismo de ácidos grasos, como hígado, tejido adiposo marrón, riñón, corazón y músculo esquelético.12,13 Los PPARδ/β se expresan de forma ubicua y los PPARγ tienen un patrón de expresión restringido, principalmente en tejido adiposo blanco y marrón, mientras que otros tejidos como el músculo esquelético y el corazón contienen cantidades limitadas.12 Para ser transcripcionalmente activos, los PPAR necesitan heterodimerizar con el receptor del ácido 9-cis retinoico (RXR) (NR2B). Los heterodímeros PPAR-RXR se unen a secuencias específicas de ADN llamadas elementos de respuesta a proliferadores de peroxisomas (peroxisome proliferator response element [PPRE]), que consisten en dos repeticiones AGGTCA separadas por un único nucleótido (Direct Repeat 1 [DR-1]) (figura 1).

Figura 1. Mecanismo de acción transcripcional de las TZD.
Estas secuencias han sido caracterizadas dentro de regiones promotoras de genes diana de PPAR. En ausencia del ligando, se forman complejos de alta afinidad entre los heterodímeros PPAR-RXR y las proteínas correpresoras del receptor nuclear, las cuales evitan la activación transcripcional por secuestro del heterodímero desde el promotor. La unión del ligando a PPAR induce un cambio conformacional dando lugar a una disociación de las proteínas correpresoras, y entonces el heterodímero puede unirse a los PPRE. Además, una vez que el heterodímero es activado por el ligando, es capaz de reclutar proteínas coactivadoras que provocan el inicio de la transcripción. Como consecuencia de estos cambios en la actividad transcripcional, la unión de ligandos al receptor conlleva cambios en los niveles de expresión de ARNm de los genes diana de PPAR. En un determinado contexto celular son muchos los factores que regulan la actividad transcripcional de los PPAR (expresión relativa de los PPAR, el contexto del promotor de los genes diana, la presencia de proteínas coactivadoras y correpresoras, etc.).
Los PPAR también son capaces de regular la expresión genética independientemente de la unión a los PPRE. Pueden interactuar físicamente con otros tipos de factores de transcripción e influir en su función sin unirse al ADN, mediante un mecanismo denominado transrepresión dependiente de receptor.14 A través de este mecanismo independiente de la unión al ADN se suprime la actividad de diversos factores de transcripción, incluidos NF-κB, AP-1 y STAT-1. La mayoría de los efectos antiinflamatorios de los PPAR son explicados probablemente por este mecanismo.15 Además, recientemente se demostró que la inhibición de la actividad NF-κB es un mecanismo por el cual los agonistas de PPARγ mejoran la sensibilidad a la insulina in vivo y que el NF-κB adipocitario es una diana terapéutica potencial para la obesidad y la diabetes de tipo 2.16
Existen tres mecanismos principales de transrepresión por los cuales los complejos PPAR-RXR activados por ligando pueden regular negativamente las actividades de otros factores de transcripción. En el primero, la transrepresión es consecuencia de una competición por cantidades limitadas de coactivadores compartidos. Bajo estas condiciones en las cuales los niveles de coactivadores específicos son el factor limitante, la activación de PPAR suprime la actividad de otros factores de transcripción que utilizan el mismo coactivador.17,18 En el segundo mecanismo se cree que los heterodímeros PPAR-RXR activados actúan a través de interacciones físicas con otros factores de transcripción (por ejemplo AP-1, NF-κB, NFAT o STAT). Esta asociación impide la unión del factor de transcripción a su elemento de respuesta y por tanto inhibe su capacidad de inducir la transcripción génica.19,20 El último mecanismo de transrepresión consiste en la inhibición de la fosforilación y en la activación de ciertos miembros de la cascada MAPK (mitogen-activated protein kinase),21 por los heterodímeros PPAR-RXR, evitando así la activación de factores de transcripción.
Las TZD fueron la primera clase de compuestos identificados como ligandos de PPARγ. La implicación de PPARγ en los efectos farmacológicos de las TZD se basó en estudios que mostraban excelente correlación entre la acción hipoglucemiante de estos fármacos y su afinidad por PPARγ.22 A pesar de dicha evidencia, el lugar de acción y el mecanismo molecular de las TZD aún no están claros. Aunque el músculo esquelético es capaz de utilizar hasta un 80% de la glucosa,23 se cree que el lugar primario de la acción de las TZD es el tejido adiposo. Esta asunción está basada en los niveles existentes de PPARγ en tejido adiposo, que sobrepasan de 20 a 30 veces los existentes en músculo o hígado.24-26 A pesar de los bajos niveles de expresión de PPARγ en músculo, diversos estudios sugieren un efecto directo de las TZD en este tejido. Recientemente se llevaron a cabo estudios en diferentes modelos animales para intentar explicar el hecho de que fármacos antidiabéticos como las TZD ejerzan su acción promoviendo la adipogénesis cuando el factor de riesgo mayoritario para el desarrollo de la diabetes tipo 2 es la obesidad. El objetivo de este artículo es revisar los estudios más relevantes llevados a cabo para intentar establecer el mecanismo de acción de las TZD antidiabéticas.
Estudio del mecanismo de acción de las tiazolidinedionas antidiabéticas mediante el uso de modelos animales
Los agonistas PPARγ se utilizaron en diferentes animales obesos y diabéticos con resistencia a la insulina.27-32 La administración de estos fármacos en modelos animales con diabetes disminuyó los niveles de glucosa y de insulina plasmáticos, incrementando por tanto la sensibilidad a la insulina. De hecho, en modelos animales con resistencia a la insulina, como las ratas obesas Zucker,27 ratas obesas y diabéticas Zucker,28 ratas sometidas a una dieta con fructosa,29 ratas tratadas con TNFα,30 ratas tratadas con glucosamina31 y ratas alimentadas con una dieta grasa,32 el tratamiento con las TZD incrementó la sensibilidad a la insulina y, por tanto, provocó un aumento en la utilización de glucosa.33,34 Los efectos hipoglucemiantes de estos fármacos se correlacionaron fuertemente con su afinidad por PPARγ y con su potencia en estudios de lipogénesis.
El hecho de que las TZD lleven a cabo sus efectos antidiabéticos mediante una activación de PPARγ plantea diferentes contradicciones referentes al lugar de acción y al mecanismo molecular de estos fármacos. La primera, ¿cómo estos fármacos que activan un receptor expresado principalmente en el tejido adiposo aumentan la disponibilidad de glucosa en el músculo esquelético,35 siendo éste el tejido más sensible a la insulina No está del todo claro si las TZD producen efectos sensibilizadores a la insulina mediante cambios en el tejido adiposo o si los efectos directos de estos fármacos en el músculo esquelético y otros tejidos diferentes del adiposo son suficientes para afectar la homeostasis de la glucosa.
Hay estudios que apoyan ambas posibilidades. La implicación del tejido adiposo en la acción antidiabética de las TZD fue estudiada por diversos grupos utilizando modelos animales de lipoatrofia. En uno de estos modelos, que presentaba tejido adiposo residual e hiperinsulinemia grave, hiperglucemia e hiperlipidemia, la administración de troglitazona mejoró la sensibilidad a la insulina, normalizó la tolerancia a la glucosa y redujo los niveles de insulina y lípidos plasmáticos.36 Estos resultados sugerían que la acción antidiabética de las TZD podía producirse por vías independientes del adipocito. Este hecho coincidía con la capacidad de las TZD para intensificar el transporte de glucosa en las células de cultivos musculares, un efecto secundario directo de estos fármacos en tejidos no adiposos.37 Por el contrario, en un modelo de ratón con lipoatrofia más grave (ratón A-ZIP/F-1), modelo que no presenta tejido adiposo visible, con resistencia a la insulina e hiperlipidemia graves, el tratamiento con TZD redujo los niveles de lípidos plasmáticos pero no afectó los niveles de insulina ni los de glucosa.38 Estos resultados indican que mientras los efectos hipolipidémicos de las TZD en estos ratones eran independientes de la grasa, sus efectos hipoglucemiantes eran dependientes de la presencia de tejido adiposo. No está claro por qué estos dos modelos de animales con lipoatrofia responden de forma diferente a las TZD, pero la diferencia en la intensidad de la lipoatrofia parece estar implicada. Especialmente interesante resulta el hecho de que el trasplante de grasa en los ratones A-ZIP/F-1 normalizara la recaptación de glucosa y revirtiera la resistencia a la insulina, lo cual sugiere que son necesarios ciertos niveles de grasa para mantener los niveles de glucosa normales.38-40 En un estudio reciente se demostró que el componente del tejido adiposo que permite la normalización de la homeostasis de glucosa en los ratones A-ZIP/F-1 es la leptina.41 La leptina es una hormona derivada del adipocito que juega un papel importante en el control del gasto de energía y de la ingesta de comida.42,43 Los niveles de leptina plasmáticos se correlacionan con el grado de adiposidad44,45 y esta deficiencia en ratones causa diabetes y resistencia a la insulina.46,47 Ebihara y col. (2001) cruzaron ratones A-ZIP/F-1 y ratones transgénicos que sobreexpresaban leptina, para generar un ratón doblemente transgénico deficiente en tejido adiposo pero con elevados niveles de leptina. Este ratón doblemente transgénico mostraba una mejoría en la resistencia a la insulina y la diabetes en comparación con los ratones A-ZIP/F-1. Este resultado sugiere que los efectos antidiabéticos de las TZD en el músculo esquelético requieren la presencia de diversos factores que son secretados por el tejido adiposo.
Para intentar identificar si los efectos de las TZD sobre el músculo eran directos o indirectos, vía tejido adiposo, Norris y col. (2003) generaron ratones deficientes en PPARγ en músculo (MuPPARγKO) utilizando recombinación Cre/loxP. Los autores demostraron que estos ratones presentaban un exceso de tejido adiposo, a pesar de una reducción en la ingesta de dieta, y resistencia a la insulina. La sensibilidad a la insulina en el músculo esquelético de estos ratones era normal pero estaba alterada en el hígado. Aunque los ratones MuPPARγKO mostraban alterada la expresión de diversos genes del metabolismo lipídico en el músculo respondían normalmente a la rosiglitazona. Estos datos sugerían que los PPARγ eran críticos para el mantenimiento de la sensibilidad a la insulina en el músculo, pero no eran necesarios para las acciones antidiabéticas de las TZD. Serán necesarios futuros estudios para aclarar definitivamente el lugar de acción de las TZD.
La segunda contradicción aparente se refiere al hecho de que siendo la obesidad el mayor factor de riesgo para el desarrollo de la diabetes tipo 2, ¿cómo las TZD, que promueven la adipogénesis, pueden mejorar la resistencia a la insulina De hecho, la administración de TZD en roedores produce un incremento en los depósitos de grasa como resultado de la diferenciación de adipocitos mediada por PPARγ.48 Se cree que la administración de TZD mejora la resistencia a la insulina debido a su capacidad de estimular la diferenciación adipocitaria al generar más adipocitos, los cuales son más pequeños en tamaño. En la diabetes tipo 2, los adipocitos son más grandes que en individuos normales. Estos adipocitos más grandes secretan elevados niveles de ácidos grasos libres y del factor de necrosis tumoral α (TNFα), los cuales se relacionan con la inducción de resistencia a la insulina en hígado y en músculo esquelético.49,50 Los adipocitos pequeños generados por las TZD son más sensibles a la insulina y liberan menos ácidos grasos libres y TNFα. Por tanto, la proliferación de adipocitos pequeños y la disminución del número de adipocitos grandes en el tejido adiposo después de la administración de TZD pueden proporcionar una explicación del mecanismo molecular de estos fármacos antidiabéticos. Para profundizar en el papel de los PPARγ en la homeostasis de la glucosa y la resistencia a la insulina, una de las estrategias seguidas fue la generación de ratones con disrupción dirigida del gen de este receptor. Los ratones homocigotos PPARγ-/- no son viables, pero los ratones heterocigotos PPARγ+/-, que presentan una reducción del 50% en la expresión de PPARγ, son un modelo animal excelente para estudiar el papel de este receptor nuclear.51,52 Basados en los efectos observados después de la activación de PPARγ por las TZD, se esperaba que los ratones que habían perdido un alelo PPARγ reducirían su sensibilidad a la insulina. No obstante, cuando estos animales fueron expuestos a una dieta con elevado contenido de grasas, presentaron menor aumento de peso y resistencia a la insulina respecto de ratones normales.53 Estos sorprendentes resultados fueron justificados por los bajos niveles de PPARγ presentes en los adipocitos de estos ratones heterocigotos, lo que causa una disminución en la diferenciación adipocitaria al generar adipocitos más pequeños y conducir a un aumento de la sensibilidad a la insulina. Además, estos ratones presentan niveles elevados de leptina circulante, como consecuencia de la inhibición de la transcripción del gen de la leptina por PPARγ,54 y como resultado de ello muestran una disminución en la ingesta de comida y una tasa metabólica basal más alta. Estos datos inesperados plantearon la cuestión de si los PPARγ favorecían o evitaban la obesidad y la resistencia a la insulina. Por una parte, la activación de PPARγ por las TZD genera adipocitos pequeños, más sensibles a la insulina, dando lugar a una mejora de la resistencia a la insulina. No obstante, la activación excesiva de PPARγ incrementa los depósitos de grasa y reduce la producción de leptina55 dando lugar a hiperfagia y resistencia a la insulina. Por otro lado, los ratones heterocigotos PPARγ+/- están protegidos contra la obesidad y la resistencia a la insulina. Esta última posibilidad se investigó posteriormente al tratar ratones con antagonistas de PPARγ (BADGE) y RXR (HX531).56 En ratones normales, el antagonismo de PPARγ/RXR potenció el efecto de la leptina y aumentó la combustión de los ácidos grasos y la disipación de energía, mejorando así la obesidad inducida por una dieta rica en grasas y la resistencia a la insulina. Por el contrario, el tratamiento de los ratones heterocigotos PPARγ+/- con estos antagonistas dio lugar a reducción del tejido adiposo blanco y a una marcada disminución en los niveles de leptina y la disipación de energía, dando lugar a una reaparición de resistencia a la insulina. Basados en estos datos, Yamauchi y col. (2001) propusieron una posible hipótesis según cual PPARγ/RXR regularía la sensibilidad a la insulina. Según estos autores, los niveles óptimos de la actividad PPARγ/RXR para mantener la sensibilidad a la insulina son 0.3 a 0.5 veces la actividad normal. Un incremento en la actividad de PPARγ/RXR parece producir una reducción en los niveles de leptina, ya que este receptor inhibe la transcripción del gen de la leptina,54 y promueve el almacenamiento de grasa, causando resistencia a la insulina. Por el contrario, una reducción de la actividad PPARγ/RXR inferior a los niveles óptimos de 0.3 a 0.5 conllevaría una disminución de los niveles de leptina circulantes debido a la desaparición del tejido adiposo. Por tanto, una activación excesiva o insuficiente de PPARγ/RXR podría producir un deterioro de la vía de la leptina, causando una reducción en la oxidación de ácidos grasos en hígado y músculo esquelético que, finalmente, conduciría a resistencia a la insulina. Evidencias genéticas sugieren que PPARγ actúa como un gen ahorrativo de energía.53,57 Por tanto, PPARγ puede ser considerado como un gen que favorece el almacenamiento de grasa durante las etapas de escasez de alimentos al incrementar la supervivencia durante estos períodos. No obstante, en ciclos de ingesta calórica excesiva, típica de las sociedades industrializadas, PPARγ predispondría a sufrir obesidad y diabetes tipo 2.3
Estudios recientes también parecen indicar que las TZD pueden llevar a cabo sus acciones al evitar la reducción de genes del metabolismo oxidativo durante el desarrollo de la resistencia a la insulina, alteración implicada en la patogénesis de la diabetes mellitus tipo 2.58 El músculo esquelético es responsable de la utilización de la mayor parte de la glucosa y es, por tanto, el lugar mayoritario de resistencia a la insulina en la obesidad y en la diabetes mellitus tipo 2.59 Durante el desarrollo de la resistencia a la insulina en el músculo esquelético se observa un cambio en la fuente de sustratos energéticos, la utilización de glucosa se reduce y aumenta la de ácidos grasos. El transporte y el metabolismo de estos sustratos son controlados por los PPAR. En las células del músculo esquelético la utilización de ácidos grasos y la expresión de diversos genes implicados en la β-oxidación mitocondrial de los ácidos grasos se encuentra bajo control de PPARα.60 Además, la expresión de la proteína de PPARα es inducida durante la diferenciación del miocito, lo que coincide con un aumento en la capacidad de β-oxidación de los ácidos grasos.60 La β-oxidación mitocondrial de estos ácidos grasos da lugar a equivalentes reductores en forma de NADH, los cuales son oxidados por la cadena de transporte electrónico (CTE), con el oxígeno como aceptor final del transporte electrónico (respiración) (figura 2).
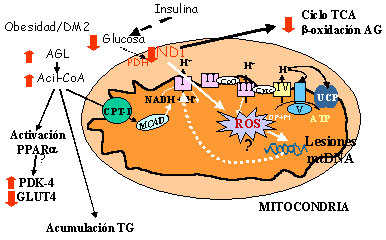
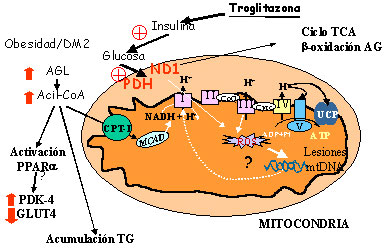
Figura 2. En la cadena de transporte electrónico (CTE) mitocondrial, 6 subunidades del complejo I (subunidades 1-6 de la NADH deshidrogenasa, [ND1-6]), 1 subunidad del complejo III (citocromo b, [cytb]), 3 subunidades del complejo IV (citocromo c oxidasa I-III, [COI-III]) y dos subunidades del complejo V (subunidades 6 y 8 de la ATP sintasa) son codificadas por el ADN mitocondrial (mtDNA). La reducción de la actividad de la CTE mitocondrial provoca estrés oxidativo debido a la acumulación de electrones en las primeras etapas de la CTE (complejo I y coenzima Q), donde estos electrones pueden ser donados directamente al oxígeno molecular para formar el anión superóxido. Una reducción en la expresión de genes implicados en el sistema de transporte mitocondrial, como por ejemplo ND1, reduciría su actividad conduciendo a diferentes trastornos mitocondriales. Durante el desarrollo de resistencia a la insulina en el músculo esquelético se observa un cambio en la fuente de sustratos energéticos, la utilización de glucosa se reduce y aumenta la de ácidos grasos. La menor utilización de glucosa durante la el desarrollo de la resistencia a la insulina podría ser responsable, al menos en parte, de la reducción en la expresión de los genes codificados por el mtDNA. El tratamiento con troglitazona revierte estas reducciones en la expresión de genes codificados por el mtDNA que participan en la CTE en estados diabéticos. Este efecto de la troglitazona se atribuye a su capacidad para incrementar la sensibilidad a la insulina y la utilización de glucosa por el músculo. Abreviaturas: AGL, ácidos grasos libres; CPT-I, carnitina palmitoiltransferasa I; GLUT4, transportador de glucosa; ND1, subunidad 1 de la NADH deshidrogenasa (complejo I de la cadena de transporte electrónico mitocondrial); PDK-4, piruvato deshidrogenasa quinasa 4; PDH, piruvato deshidrogenasa; ROS, especies reactivas de oxígeno; TCA, ciclo de los ácidos tricarboxílicos; TG, triglicéridos.
Los complejos I, III y IV de la CTE, pero no el complejo II, acoplan el transporte electrónico a la bomba de protones creando un gradiente electroquímico. Este gradiente de protones es entonces utilizado para sintetizar ATP por el complejo V, en un proceso conocido como fosforilación oxidativa. Es interesante destacar que 6 subunidades del complejo I (subunidades 1-6 de la NADH deshidrogenasa [ND1-6]), 1 subunidad del complejo III (citocromo b [cytb]), 3 subunidades del complejo IV (citocromo c oxidasa I-III [COI-III]) y 2 subunidades del complejo V (subunidades 6 y 8 de la ATP sintasa) son codificadas por el ADN mitocondrial (mtDNA). La reducción de la actividad de la CTE mitocondrial provoca estrés oxidativo debido a la acumulación de electrones en las primeras etapas de la CTE (complejo I y coenzima Q), donde estos electrones pueden ser donados directamente al oxígeno molecular para formar el anión superóxido.61,62 Además, la CTE está ligada a la β-oxidación mitocondrial y la inhibición de la primera da lugar a una inhibición de la segunda.63 Por lo tanto, una alteración de la expresión o de la actividad tanto de PPARα como de la CTE podría ser uno de los mecanismos implicados en la acumulación de lípidos en el músculo esquelético, que es el factor que se correlaciona más fuertemente con la resistencia a la insulina.64,65
Al intentar identificar si la alteración de la expresión de estos genes estaba implicada en la resistencia a la insulina en el músculo, estudiamos su expresión en el músculo esquelético de ratas Zucker diabetic fatty (ZDF), un modelo animal de obesidad y diabetes de tipo 2, antes y después de un tratamiento con troglitazona, fármaco que incrementa la sensibilidad a la insulina.66 En el músculo sóleo de ratas ZDF, comparadas con ratas no obesas (lean), se observó un aumento en la expresión de PPARα y de sus genes diana. Además, la expresión de dos genes codificados por el mtDNA implicados en la CTE (ND1 y COII) estaba reducida, mientras que la expresión del gen del complejo II codificado por el ADN nuclear no resultó afectada. También se observó correlación inversa entre la reducción en la expresión de ND1 y COII en el músculo esquelético y los niveles plasmáticos de glucosa, lo cual sugiere que la reducción en la utilización de glucosa por parte del músculo esquelético estaba implicada en los cambios en la expresión de estos genes.
Además, los niveles de ARNm del coactivador de PPARγ-1 (PGC-1), que está implicado en la biogénesis y la respiración mitocondrial, estaban reducidos en el músculo esquelético de las ratas ZDF. El tratamiento con troglitazona restableció la expresión de ND1 y PGC-1 en el músculo esquelético de ratas ZDF.
Estos resultados demuestran que los estados diabéticos van acompañados de una reducción de la expresión del ND1, codificada por el mtDNA, en el músculo esquelético de ratas ZDF. Este hecho es interesante puesto que Lee y col.67 demostraron que una disminución del número de copias del mtDNA precedía al desarrollo de la diabetes tipo 2, sugiriendo que reducciones en la expresión de genes codificados por el mtDNA podrían estar relacionadas con la patogénesis de esta enfermedad. La importancia del mtDNA en el desarrollo de la diabetes mellitus se debe al hecho de que distintas mutaciones del mtDNA se asocian frecuentemente con la diabetes. Así, por ejemplo, mutaciones en ND1 causan diabetes como fenotipo prominente.68 Una reducción en la expresión de genes implicados en el sistema de transporte mitocondrial como la comentada podría reducir su actividad y conducir a diferentes trastornos mitocondriales. Por ejemplo, la supresión del sistema de transporte electrónico se relacionó con la desaceleración del ciclo del ácido tricarboxílico.69 Además, la alteración de la CTE reduce la β-oxidación de los ácidos grasos, puesto que el primero ejerce control sobre el segundo en diferentes puntos.63 Por tanto, la alteración de la expresión de la CTE puede ser uno de los mecanismos fundamentales en los trastornos metabólicos del músculo esquelético en la diabetes mellitus tipo 2. A pesar de todo, serán necesarios más estudios para verificar esta hipótesis.
Además, estos estudios relacionan la diabetes con cambios en la expresión de PGC-1, un coactivador para muchos factores en la familia de receptores hormonales nucleares que se relacionó con la biogénesis, respiración y termogénesis mitocondrial.70-75 El PGC-1 también interactúa físicamente con el factor intensificador de miocitos 2C (MEF2C) para aumentar la expresión de GLUT4 y la recaptación de glucosa en células L6 que sobreexpresaban PGC-1.76 Por lo tanto, la reducción de los niveles de PGC-1 en el músculo esquelético de ratas ZDF puede reducir la captación de glucosa, contribuyendo, al menos en parte, a la disminución de la expresión de genes codificados por el mtDNA.
Además, se demostró que el flujo de la insulina y de la glucosa regula la expresión de los genes codificados por el mtDNA, como ND1, en el músculo esquelético.77 Así, la infusión de insulina incrementó los niveles de ARNm de ND1 aproximadamente 2.5 veces en el músculo esquelético y la respuesta de ND1 a la insulina se correlacionó con la recaptación de glucosa.77 De forma similar, Kanazawa y col.78 observaron una reducción en la expresión de los genes codificados por el mtDNA (subunidad 6 de la ATP sintasa y el citocromo b) en corazón de ratas con diabetes inducida por estreptozotocina. El tratamiento con insulina restableció completamente la expresión de estos genes. Nuestro grupo demostró que la reducción de la expresión de ND1 y COII estaba inversamente correlacionada con los niveles de glucosa, lo que indica que la disminución en la utilización de glucosa es responsable en parte de la expresión reducida de estos genes codificados por el mtDNA. El hecho de que el tratamiento con troglitazona restableciera la expresión de ND1 y PGC-1 debería ser atribuido a una mejora de la sensibilidad a la insulina en el músculo esquelético. De acuerdo con dicha mejoría observamos reducción de la expresión de la piruvato deshidrogenasa kinasa 4 (PDK-4). Un aumento de la expresión de PDK-4 se asoció con incremento del suministro, con oxidación de ácidos grasos en el músculo esquelético o con ambos fenómenos.70 En coincidencia con este resultado, se observó incremento de los niveles de ARNm de PDK-4 en el músculo esquelético de ratas con diabetes inducida por estreptozotocina, incremento que fue revertido por el tratamiento con insulina.79 Igualmente, un aumento en la expresión de PDK-4 y una reducción en la actividad de la piruvato deshidrogenasa se asoció con menor producción de ATP.80 Tal defecto perjudicaría la síntesis de ATP, comprometiendo el reclutamiento de GLUT4, proceso dependiente de energía, a la superficie celular. Por tanto, la reducción de la expresión de PDK-4 tras el tratamiento con troglitazona mejoraría la utilización de glucosa.
Recientemente, Patti y col. demostraron que en el músculo esquelético de pacientes con diabetes tipo 2 se produce una reducción en la expresión de PGC-1.58 Así, nuestros resultados confirman que las ratas ZDF son un buen modelo animal de diabetes mellitus tipo 2 para el estudio de los cambios en la expresión genética en el músculo esquelético y, lo más importante, que el tratamiento con troglitazona restablece la expresión de PGC-1 en el músculo. Este último punto es particularmente interesante ya que la detección e interrupción de los trastornos mitocondriales contribuiría a prevenir la aparición de diabetes.
En resumen, si se confirma que la alteración de la función mitocondrial debida a reducción de la expresión de genes implicados en la CTE es uno de los mecanismos esenciales en los trastornos metabólicos del músculo esquelético en la diabetes mellitus tipo 2, un aumento en la expresión de ND1 y PGC-1 después del tratamiento con troglitazona lograría evitar dichas alteraciones.
Son necesarios nuevos estudios para aclarar el mecanismo de acción molecular de las TZD, pero está claro que el descubrimiento de estos fármacos incrementó nuestro conocimiento sobre la patogénesis de la diabetes tipo 2 y proporcionó nuevas vías para el diseño de fármacos para el tratamiento de esta enfermedad.
El autor principal manifiesta que no hay conflicto de intereses con respecto al contenido de este artículo y no ha habido apoyo económico ni financiación para la realización de su trabajo, así como otras relaciones de tipo económico o personal relativas al mismo.
Bibliografía del artículo
- Saltiel AR, Olefsky JM. Thiazolidinediones in the treatment of insulin resistance and Type II diabetes. Diabetes 1996, 45: 1661-1669.
- Spiegelman BM. PPARγ: adipogenic regulator and thiazolidinedione receptor. Diabetes 1998, 47: 507-514.
- Lowell BB. PPARγ: an essential regulator of adipogenesis and modulator of fat cell function. Cell 1999, 99: 230-253.
- Huli B, McCarthy PA, Gibbs EM. The glitazone family of antidiabetic agents. Curr. Pharm. Des. 1996, 2: 85-102.
- Ibrahimi A, Teboul L, Gaillard D y col. Evidence for a common mechanism of action for fatty acids and thiazolidinedione antidiabetic agents on gene expression in preadipose cells. Mol. Pharmacol. 1994, 46: 1070-1076.
- Kletzien RF, Clarke SD, Ulrich RG. Enhancement of adipocyte differentiation by an insulin-sensitizing agent. Mol. Pharmacol. 1992, 41: 393-398.
- Kletzien RF, Foellmi LA, Harris PK y col. Adipocyte fatty acid-binding protein: regulation of gene expression in vivo and in vitro by an insulin-sensitizing agent. Mol. Pharmacol. 1992, 42: 558-562.
- Tontonoz P, Hu E, Spiegelman BM. Stimulation of adipogenesis in fibroblasts by PPARγ2, a lipid-activated transcription factor. Cell 1994, 79: 1147-1156.
- Forman BM, Tontonoz P, Chen J y col. 15-Deoxy-Δ12,14-prostaglandin J2 is a ligand for the adipocyte determination factor PPARγ. Cell 1995, 83: 803-812.
- Lehmann JM, Moore LB, Smith-Oliver y col. An antidiabetic thiazolidinedione is a high affinity ligand for peroxisome proliferator-activated receptor γ (PPARγ). J. Biol. Chem. 1995, 270: 12953-12956.
- A unified nomenclature system for the nuclear receptor superfamily. Cell 1999, 97:161-163.
- Braissant O, Foufelle F, Scotto C y col. Differential expression of peroxisome proliferator-activated receptors (PPAR): tissue distribution of PPAR-alpha, -beta, and -gamma in the adult rat. Endocrinology 1996, 137: 354-366.
- Desvergne B, Wahli W. Peroxisome proliferator-activated receptors: nuclear control of metabolism. Endocr. Rev. 1999, 20:649-688.
- Chinetti G, Fruchart JC, Staels B. Peroxisome proliferator-activated receptors (PPAR): nuclear receptors at the crossroads between lipid metabolism and inflammation. Inflamm. Res. 2000, 49:497-505.
- Daynes RA, Jones DC. Emerging roles of PPAR in inflammation and immunity. Nat. Rev. Immunol. 2002, 2: 748-759.
- Ruan H, Pownall HJ, Lodish HF. Troglitazone antagonizes tumor necrosis factor-alpha-induced reprogramming of adipocyte gene expression by inhibiting the transcriptional regulatory functions of NF-kappaB. J. Biol. Chem. 2003, 278:28181-28192.
- Li M, Pascual G, Glass CK. Peroxisome proliferator-activated receptor gamma-dependent repression of the inducible nitric oxide synthase gene. Mol. Cell. Biol. 2000, 20:4699-46707.
- Kamei Y, Xu L, Heinzel T y col. A CBP integrator complex mediates transcriptional activation and AP-1 inhibition by nuclear receptors. Cell 1996, 85:403-414.
- Delerive P, De Bosscher K, Besnard S y col. Peroxisome proliferator-activated receptor alpha negatively regulates the vascular inflammatory gene response by negative cross-talk with transcription factors NF-kappaB and AP-1. J. Biol. Chem. 1999, 274:32048-32054.
- Delerive P, Martin-Nizard F, Chinetti G y col. Peroxisome proliferator-activated receptor activators inhibit thrombin-induced endothelin-1 production in human vascular endothelial cells by inhibiting the activator protein-1 signaling pathway. Circulation Res. 1999, 85:394-402.
- Desreaumaux P, Dubuquoy L, Nutten S y col. Attenuation of colon inflammation through activators of the retinoid X receptor (RXR)/peroxisome proliferator-activated receptor gamma (PPARgamma) heterodimer. A basis for new therapeutic strategies. J. Exp. Med. 2001, 193:827-838.
- Willson TM, Cobb JE, Cowan DJ y col. The structure-activity relationship between peroxisome-proliferator activated receptor γ agonism and the antihyperglycemic activity of thiazolidinediones. J. Med. Chem. 1996, 39: 665-668.
- Olefsky JM. Treatment of insulin resistance with peroxisome proliferator-activated receptor agonists. J. Clin. Invest. 2000, 106: 467-472.
- Kruszynska YT, Mukherjee R, Jow L y col. Skeletal muscle peroxisome proliferator-activated receptor-γ expression in obesity and non-insulin-dependent diabetes mellitus. J. Clin. Invest. 1998, 101: 543-548.
- Vidal-Puig AJ, Considine RV, Jimenez-Linan M, y col. Peroxisome proliferator-activated receptor gene expression in human tissues: effects of obesity, weight loss, and regulation by insulin and glucocorticoids. J. Clin. Invest. 1997, 99: 2416-2422.
- Park KS, Ciaraldi TP, Abrams-Carter L y col. PPARγ gene expression is elevated in skeletal muscle of obese and type II diabetic subjects. Diabetes 1997, 46: 1230-1234.
- Fujiwara T, Yoshioka S, Yoshioka T y col. Characterization of CS-045, a new oral antidiabetic agent. II. Effects on glycemic control and pancreatic islet structure at a late stage of the diabetic syndrome in C57BL/Ksj-db/db mice. Metabolism 1991, 40: 1213-1218.
- Fujiwara T, Yoshioka S, Yoshioka T y col. Characterization of new oral antidiabetic agent CS-045: studies in KK and ob/ob mice and Zucker fatty rats. Diabetes 1988, 37: 1549-1558.
- Lee MK, Miles PD, Khoursheed M y col. Metabolic effects of troglitazone on fructose-induced insulin resistance in the rat. Diabetes 1994, 43: 1435-1439.
- Miles PD, Romeo OM, Higo K y col. TNFα-induced insulin resistance in vivo and its prevention by troglitazone. Diabetes 1997, 46: 1678-1683.
- Miles PG, Higo K, Romeo OM y col. Troglitazone prevents hyperglycemia-induced but not glucosamin-induced insulin resistance. Diabetes 1998, 47: 395-400.
- Kraegen EW, James DE, Jenkins AB y col. A potent in vivo effect of ciglitazone on muscle insulin resistance induced by high fat feeding of rats. Metabolism 1989, 38: 1089-1093.
- Arakawa K, Inamasu M, Matsumoto M y col. Novel benzoxazole 2,4-thiazolidinediones as potent hypoglycemic agents. Synthesis and structure-activity relationships. Chem. Pharm. Bull. 1997 45:1984-1993.
- Reginato MJ, Bailey ST, Krakow SL y col. A potent antidiabetic thiazolidinedione with unique peroxisome proliferator-activated receptor gamma-activating properties. J. Biol. Chem. 1998, 273: 32679-32684
- Inzucchi SE, Maggs DG, Spollett GR, Page SL y col. Efficacy and metabolic effects of metformin and troglitazone in type II diabetes mellitus. N. Engl. J. Med. 1998, 338: 867-872.
- Burant CF, Sreenen S, Hirano KI y col. Troglitazone action is independent of adipose tissue. J. Clin. Invest. 1997, 100: 2900-2908.
- Ciaraldi TP, Gilmore A, Olefsky JM y col. In vitro studies on the action of CS-045. A new anti-diabetic agent. Metabolism 1990, 39: 1056-1062.
- Chao L, Marcus-Samuels B, Mason MM y col. Adipose tissue is required for the antidiabetic, but not for the hypolipidemic, effect of thiazolidinediones. J. Clin. Invest. 2000, 106: 1221-1228.
- Kim JK, Gavrilova O, Chen Y y col. Mechanism of insulin resistance in A-ZIP/F-1 fatless mice. J. Biol. Chem. 2000, 275: 8456-8460.
- Gavrilova O, Marcus-Samuels B, Graham D y col. Surgical implantation of adipose tissue reverses diabetes in lipoatrophic mice. J. Clin. Invest. 2000, 105: 271-278.
- Ebihara K, Ogawa Y, Masuzaki H y col. Transgenic overexpression of leptin rescues insulin resistance and diabetes in a mouse model of lipoatrophic diabetes. Diabetes 2001, 50: 1440-1448.
- Zhang Y, Proenca R, Maffei M y col. Positional cloning of the mouse obese gene and its human homologue. Nature 1994, 372: 425-432.
- Ogawa Y, Masuzaki H, Isse N y col. Molecular cloning of rat obese cDNA and augmented gene expression in genetically obese Zucker fatty (fa/fa) rat. J. Clin. Invest. 1996, 96: 1280-1287.
- Maffei H, Hallas J, Ravussin E y col. Leptin levels in human and rodent: measurement of plasma leptin and ob RNA obese and weight-reduced subjects. Nature Med. 1995, 1: 1155-1161.
- Considine RV, Sinha MK, Heiman ML y col. Serum immunoreactive-leptin concentrations in normal-weight and obese humans. N. Engl. J. Med. 1996, 334: 292-295.
- Montague CT, Farooqi IS, Whitehead JP y col. Congenital leptin deficiency is associated with severe early-onset obesity in humans. Nature 1997, 387: 903-908.
- Strobel A, Issad T, Camoin L y col. A leptin missense mutation associated with hypogonadism and morbid obesity. Nat. Gent. 1998, 18: 213-215.
- Schoonjans K, Auwerx J. Thiazolidinediones: an update. Lancet 2000 356:254-255.
- Hotamisligil GS, Peraldi P, Spiegelman BM. The molecular link between obesity and diabetes. Curr. Opin. Endocrinol. Diabetes 1996, 3: 16-23.
- Rebrin K, Steil GM, Mittelman SD y col. Causal linkage between insulin supression of lipolysis and supression of liver glucose output in dogs. J. Clin. Invest. 1996, 98: 741-749.
- Barak Y, Nelson MC, Ong ES y col. PPARγ is required for placental, cardiac, and adipose tissue development. Mol. Cell. 1999, 4: 585-595.
- Miles PD, Barak Y, Evans RM y col. Improved insulin-sensitivity in mice heterozygous for PPARγ deficiency. J. Clin. Invest. 2000, 105: 287-292.
- Kubota N, Terauchi Y, Miki H y col. PPARγ mediates high-fat induced adipocyte hypertrophy and insulin resistance. Mol. Cell 1999, 4: 585-595.
- De Vos P, Lefebvre AM, Miller SG y col. Thiazolidinediones repress ob gene expression in rodents via activation of peroxisome proliferator-activated receptor γ. J. Clin. Invest. 1996, 98: 1004-1009.
- Girard J. Is leptin the link between obesity and insulin resistance Diabetes Metab. 1997, 4: 611-617.
- Yamauchi T, Waki H, Kamon J y col. Inhibition of RXR and PPARγ ameliorates diet-induced obesity and type 2 diabetes. J. Clin. Invest. 2001, 108: 1001-1013.
- Auwerx J. PPARγ, the ultimate thrifty gene. Diabetologia 1999, 42: 1033-1049.
- Patti ME, Butte AJ, Crunkhorn S y col. Coordinated reduction of genes of oxidative metabolism in humans with insulin resistance and diabetes: Potential role of PGC1 and NRF1. Proc. Natl. Acad. Sci. U.S.A. 2003, 100:8466-8471.
- DeFronzo RA, Gunnarsson R, Bjorkman O y col. Effects of insulin on peripheral and splanchnic glucose metabolism in noninsulin-dependent (type II) diabetes mellitus. J. Clin. Invest. 1985, 76:149-155.
- Muoio DM, Way JM, Tanner CJ y col. Peroxisome proliferator-activated receptor-alpha regulates fatty acid utilization in primary human skeletal muscle cells. Diabetes 2002, 51:901-909.
- Wallace DC. Mitochondrial diseases in man and mouse. Science 1999, 283:1482-1488.
- Brambilla L, Cairo G, Sestili P y col. Mitochondrial respiratory chain deficiency leads to overexpression of antioxidant enzymes. Febs Lett 1997, 418:247-250.
- Eaton S, Pourfarzam M, Bartlett K. The effect of respiratory chain impairment of beta-oxidation in rat heart mitochondria. Biochem. J. 1996, 319:633-640.
- Perseghin G, Scifo P, De Cobelli F y col. Intramyocellular triglyceride content is a determinant of in vivo insulin resistance in humans: a 1H-13C nuclear magnetic resonance spectroscopy assessment in offspring of type 2 diabetic parents. Diabetes 1999, 48:1600-1606.
- Oakes ND, Cooney GJ, Camilleri S y col. Mechanisms of liver and muscle insulin resistance induced by chronic high-fat feeding. Diabetes 1997, 46, 1768-1774.
- Jove M, Salla J, Planavila A y col. Impaired expression of the mitochondrial DNA-encoded gene NADH dehydrogenase subunit 1 and PPARgamma coactivator-1 in skeletal muscle of ZDF rats: restoration by troglitazone treatment. J. Lipid Res. 2004, 45, 113-123.
- Lee HK, Song JH, Shin CS y col. Decreased mitochondrial DNA content in peripheral blood precedes the development of non-insulin-dependent diabetes mellitus. Diabetes Res Clin Pract 1998, 42:161-167.
- Maassen JA, Janssen GM, Lemkes HH. Mitochondrial diabetes mellitus. J. Endocrinol. Invest. 2002, 25:477-484.
- Noda M, Yamashita S, Takahashi N y col. Switch to anaerobic glucose metabolism with NADH accumulation in the beta-cell model of mitochondrial diabetes. Characteristics of betaHC9 cells deficient in mitochondrial DNA transcription. J Biol Chem 2002, 277: 41817-41826.
- Puigserver P, Wu Z, Park CW y col. A cold-inducible coactivator of nuclear receptors linked to adaptive thermogenesis. Cell. 1998, 92:829-839.
- Esterbauer H, Oberkofler H, Krempler F y col. Human peroxisome proliferator activated receptor gamma coactivator 1 (PPARGC1) gene: cDNA sequence, genomic organization, chromosomal localization, and tissue expression. Genomics 1999, 62:98-102.
- Lin J, Wu H, Tarr PT y col. Transcriptional co-activator PGC-1 alpha drives the formation of slow-twitch muscle fibres. Nature 2002, 418:797-801.
- Lin J, Puigserver P, Donovan J y col. Peroxisome proliferator-activated receptor gamma coactivator 1beta (PGC-1beta ), a novel PGC-1-related transcription coactivator associated with host cell factor. J. Biol. Chem. 2002, 277:1645-1648.
- Kressler D, Schreiber SN, Knutti D y col. The PGC-1-related protein PERC is a selective coactivator of estrogen receptor alpha. J Biol Chem. 2002, 277:13918-13925.
- Wu Z, Puigserver P, Andersson U y col. Mechanisms controlling mitochondrial biogenesis and respiration through the thermogenic coactivator PGC-1. Cell 1999, 98:115-124.
- Michael LF, Wu Z, Cheatham RB y col. Restoration of insulin-sensitive glucose transporter (GLUT4) gene expression in muscle cells by the transcriptional coactivator PGC-1. Proc Natl Acad Sci U S A. 2001, 98:3820-3825.
- Huang X, Eriksson KF, Vaag A y col. Insulin-regulated mitochondrial gene expression is associated with glucose flux in human skeletal muscle. Diabetes 1999, 48: 1508-1514.
- Kanazawa A, Nishio Y, Kashiwagi A y col. Reduced activity of mtTFA decreases the transcription in mitochondria isolated from diabetic rat heart. Am. J. Physiol. Endocrinol. Metab. 2002, 282:E778-E785.
- Sugden MC, Bulmer K, Holness MJ. Fuel-sensing mechanisms integrating lipid and carbohydrate utilization. Biochem Soc Trans 2001, 29:272-278.
- Selak MA, Storey BT, Peterside I, Simmons RA. Impaired oxidative phosphorylation in skeletal muscle of intrauterine growth-retarded rats. Am J Physiol Endocrinol Metab. 2003, 285:E130-13.
©
Está
expresamente prohibida la redistribución y la redifusión de todo o parte de los
contenidos de la Sociedad Iberoamericana de Información Científica (SIIC) S.A. sin
previo y expreso consentimiento de SIIC



