

-
 Alergia
Alergia -
 Anestesiología
Anestesiología -
 Bioquímica
Bioquímica
-
 Cardiología
Cardiología
-
 Cirugía
Cirugía
-
 Dermatología
Dermatología
-
 Endocrinología y Metabolismo
Endocrinología y Metabolismo
-
 Enfermería
Enfermería
-
 Gastroenterología
Gastroenterología
-
 Hematología
Hematología
-
 Infectología
Infectología
-
 Inmunología
Inmunología
-
 Medicina Interna
Medicina Interna
-
 Nefrología
Nefrología
-
 Neumonología
Neumonología
-
 Neurología
Neurología
-
 Nutrición
Nutrición
-
 Obstetricia y Ginecología
Obstetricia y Ginecología
-
 Odontología
Odontología
-
 Oncología
Oncología
-
 Otorrinolaringología
Otorrinolaringología
-
 Pediatría
Pediatría
-
 Salud Mental
Salud Mental
-
 Salud Pública
Salud Pública
-
 Urología
Urología
-
 Más Especialidades
Más Especialidades
- Índice
-
Acceso por especialidad
-
Alergia
-
Anestesiología
-
Bioquímica
-
Cardiología
-
Cirugía
-
Dermatología
-
Endocrinología y Metabolismo
-
Enfermería
-
Gastroenterología
-
Hematología
-
Infectología
-
Inmunología
-
Medicina Interna
-
Nefrología
-
Neumonología
-
Neurología
-
Nutrición
-
Obstetricia y Ginecología
-
Odontología
-
Oncología
-
Otorrinolaringología
-
Pediatría
-
Salud Mental
-
Salud Pública
-
Urología
-
Más Especialidades
-
Noticias biomédicas
-
Fuentes informativas
- Editoriales
-
COVID-19
-
INFORMES CIENTÍFICOS
- Preguntas con Respuesta
- América Latina Investiga
- Completos revisados (full text)
-
NOTICIAS/OPINIONES
-
Expertos invitados
- Por especialidad
- de Iberoamérica
- del mundo
- Entrevistas
- Casos clínicos
-
Crónicas de Autores
- Authors’ Reports
-
Instrucciones para autores
-
Expertos Preguntan
-
Novedades
- ViASIIC
- Medicina del Dolor
-
Por especialidad
-
Iberomédica
-
Informes comentados
- Textos Completos Autorizados
- Preocupaciones Profesionales
-
Red Científica Iberoamericana (RedCIbe)
-
Conceptos Categoricos
-
Acceso por especialidad

-
Alergia
-
Anestesiología
-
Bioquímica
-
Cardiología
-
Cirugía
-
Dermatología
-
Endocrinología y Metabolismo
-
Enfermería
-
Gastroenterología
-
Hematología
-
Infectología
-
Inmunología
-
Medicina Interna
-
Nefrología
-
Neumonología
-
Neurología
-
Nutrición
-
Obstetricia y Ginecología
-
Odontología
-
Oncología
-
Otorrinolaringología
-
Pediatría
-
Salud Mental
-
Salud Pública
-
Urología
-
Más Especialidades
-
Noticias biomédicas
-
Fuentes informativas
- Editoriales
- Covid-19
-
Informes Científicos
- Preguntas con Respuesta
- América Latina Investiga
- Completos revisados (full text)
- Noticias/Opiniones
- Noticias (castellano/portugués)
- Noticias (otros idiomas)
- Expertos invitados
- Por especialidad
- de Iberoamérica
- del mundo
- Entrevistas
- Casos clínicos
-
Crónicas de Autores
- Authors’ Reports
-
Instrucciones para autores
-
Expertos Preguntan
- Novedades
- ViASIIC
- Medicina del Dolor
-
Por especialidad
-
Iberomédica
-
Informes comentados
- Textos Completos Autorizados
- Preocupaciones Profesionales
- Red Científica Iberoamericana
- Por materia
- Por fecha
-
Conceptos Categóricos

Casos Clínicos
NEUROFIBROMATOSIS TIPO 1: PSEUDOANEURISMA DE ARTERIA TEMPORAL
La neurofibromatosis es una enfermedad producida por una mutación en el cromosoma 17; se caracteriza por la aparición de tumores benignos en distintos órganos que, ocasionalmente, pueden malignizarse. El diagnóstico se efectúa por la presencia de signos característicos y puede corroborarse por medio de estudios genéticos.
Coautores
Damián Gonzalo Rutolo* Álvaro Ramírez Toncel* Marcela Barón Salgado** Fernando Andrés García*** José Sabalza Castilla*
Médico, Clínica Privada Independencia, Munro, Argentina*
Médica, Clínica Privada Independencia, Munro, Argentina**
Kinesiólogo, Clínica Privada Independencia, Munro, Argentina***
Damián Gonzalo Rutolo* Álvaro Ramírez Toncel* Marcela Barón Salgado** Fernando Andrés García*** José Sabalza Castilla*
Médico, Clínica Privada Independencia, Munro, Argentina*
Médica, Clínica Privada Independencia, Munro, Argentina**
Kinesiólogo, Clínica Privada Independencia, Munro, Argentina***
Clasificación en siicsalud
Artículos originales > Expertos del Mundo >
página /dato/casiic.php/164379
Especialidades



Artículos originales > Expertos del Mundo >
página /dato/casiic.php/164379
Especialidades
Primera edición en siicsalud
11 de marzo, 2021
11 de marzo, 2021
NEUROFIBROMATOSIS TIPO 1: PSEUDOANEURISMA DE ARTERIA TEMPORAL
(especial para SIIC © Derechos reservados)
(especial para SIIC © Derechos reservados)
Introducción
La neurofibromatosis, fue descrita en 1882 por el patólogo alemán Friedrich Daniel von Recklinghausen, es una enfermedad autosómica dominante rara, producida por una mutación en el cromosoma 17, caracterizada por la aparición de tumores en distintos órganos.
En el 50% de los casos la mutación es heredada de unos de los padres.1
Se describen cuatro tipos de neurofibromatosis.
La neurofibromatosis tipo 1 es producida por mutaciones en el gen NF1 que inhiben la neurofibromina, considerada como un supresor tumoral2 localizado en el cromosoma 17q11.2,3-5 y en un pequeño porcentaje de casos por microdeleción 17q11.6,7 En el 50% de los casos es autosómica dominante, aunque puede producirse por mutaciones de novo en NF1; la penetrancia es del 100%.8 El diagnóstico suele ser clínico y confirmado por medio de estudios genéticos.
La neurofibromatosis tipo 2 es un síndrome de predisposición a tumores, de transmisión dominante, causado por mutaciones en el gen NF2 del cromosoma 22, y se caracteriza por la aparición de múltiples schwannomas y meningiomas que afectan a ambos nervios vestibulares provocando sordera, mareos o trastornos del equilibrio. El diagnóstico se basa en estudios clínicos y por neuroimágenes; existen pruebas genéticas. El pronóstico es malo, el tratamiento es la cirugía y ocasionalmente la irradiación.
La neurofibromatosis tipo 3 se caracteriza por la existencia de múltiples schwannomas, sin afección de los nervios vestibulares. Se diagnostica generalmente en la edad adulta, afecta a menos de 1 cada 40 000 personas y se caracteriza por dolor crónico, disestesias y parestesias.
Finalmente, la neurofibromatosis tipo 6 es una enfermedad cutánea extremadamente rara, caracterizada por la presencia de máculas de color café con leche, cuyo tamaño puede variar desde unos pocos milímetros hasta más de 10 centímetros. Su etiología es desconocida, aunque algunas formas se han asociado con mutaciones del gen NF1 (17q11.2); la transmisión es autosómica dominante y su diagnóstico se basa en la presencia de más de cinco máculas, que son benignas, no necesitan tratamiento médico y algunas pueden desaparecer con la edad.
Caso clínico
Paciente de 31 años con diagnóstico de neurofibromatosis tipo 1 y cirugía estética de maxilar inferior a los 20 años, que sufrió traumatismo facial aparentemente leve. Al tercer día de producido el trauma consultó por aumento del volumen de la mitad izquierda de la cara y el cuello, secundaria a importante hematoma con compromiso de la vía aérea, por lo que se efectuó intubación orotraqueal y se instauró ventilación mecánica invasiva. Debido a la falta de complejidad fue derivado a nuestra institución, con tubo orotraqueal y dependiente de ventilación mecánica, bajo efecto de fármacos depresores del sistema nervioso central (Ramsay: -4). Se efectuó tomografía axial computarizada (TAC) y angiotomografía, que informó presencia de masa heterogénea hiperdensa voluminosa en el tejido celular subcutáneo de la hemicara izquierda, con extensión desde la región frontal hasta la mitad del cuello, extendiéndose hasta la fosa pterigomaxilar. Extensión al espacio parafaríngeo con desplazamiento de la laringe y en parte de la tráquea. Arteria carótida permeable, flujo preferencial por carótida derecha, carótida izquierda permeable, pero fina en todo su recorrido.
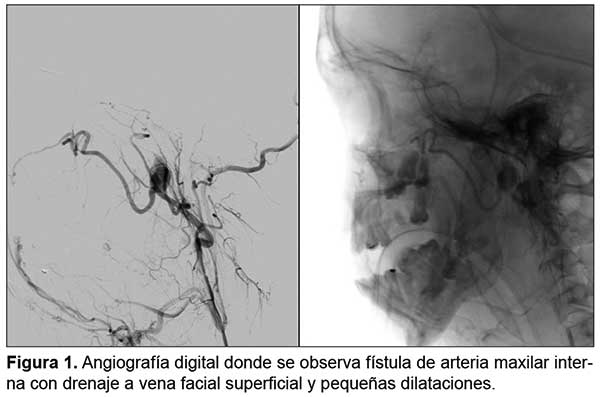
Esta masa presentaba áreas de hipervascularización y estaba irrigada por una rama de la arteria subclavia y, aparentemente, de la carótida externa. Se efectuó angiografía que mostró la presencia de un seudoaneurisma dependiente de la arteria temporal (identificándosela como responsable del sangrado), observándose por medio de la arteria maxilar interna una fístula con drenaje a la vena facial superficial y pequeñas dilataciones sin aferencias factibles de embolización, por lo que se llevó a cabo dicho procedimiento cateterizándose la rama aferente del seudoaneurisma (Figura 1).
A las 48 horas se efectuó una nueva TAC que mostró presencia de masa heterogénea, correspondiente a un hematoma, desde la región superciliar izquierda hasta la totalidad del maxilar inferior, con desviación de la tráquea hacia la derecha (Figura 2). Dado el tiempo de intubación orotraqueal (10 días) y la imposibilidad de retiro del tubo orotraqueal por inminencia de obstrucción de la vía aérea, se efectuó traqueostomía (Figura 3), con lo que el paciente fue desvinculado del ventilador sin complicaciones. Se drenaron a los 14 días del evento mediante punción guiada por ecografía, aproximadamente 350 centímetros cúbicos (Figura 4). El paciente evolucionó favorablemente, con buena mecánica ventilatoria, con ingesta de alimentos con buena tolerancia.



Discusión
La neurofibromatosis tipo 1 se produce por mutaciones en el gen NF1, lo que inhibe la neurofibromina, considerada como un supresor tumoral localizado, en el cromosoma 17q11.2 y en un pequeño porcentaje de casos por microdelección 17q11. Es autosómica dominante, aunque en el 50% de los casos se produce por mutaciones de novo en NF1; la penetrancia es del 100%. Su prevalencia aproximadamente es de 1/30003 nacidos vivos y afecta por igual a ambos sexos. Suele manifestarse en la infancia, aunque los signos pueden estar presentes desde el nacimiento, y se manifiesta por cambios en la coloración de la piel (máculas de color café con leche), pecas intertriginosas que aparecen a partir de los 5 año y, posteriormente, formación de múltiples neurofibromas cutáneos y subcutáneos que, con el transcurso de los años, se incrementan en número y tamaño. Los neurofibromas que crecen a lo largo del nervio pueden causar deformaciones, dolor o trastornos funcionales, y pueden malignizarse. Las manifestaciones oculares son los gliomas de la vía óptica y los hamartomas del iris (nódulos de Lisch). Puede presentarse osteopenia, osteoporosis, sobrecrecimiento óseo, macrocefalia, escoliosis, displasia esquelética y seudoartrosis; también puede aparecer hipertensión arterial, alteraciones vasculares, tumores intracraneales, tumores malignos de las vainas de los nervios periféricos y, ocasionalmente, convulsiones e hidrocefalia; son frecuentes los déficits cognitivos y las dificultades del aprendizaje.
El diagnóstico se efectúa con la presencia de dos o más de los siguientes signos:9 más de seis máculas de color café con leche de 5 mm en los pacientes prepúberes, y mayores de 15 mm en los pospúberes; dos o más neurofibromas de cualquier tipo o un neurofibroma plexiforme; glioma del nervio óptico; signo de Crowe: efélides axilares o inguinales; dos o más nódulos de Lisch (hamartomas del iris); displasias óseas específicas (displasia del esfenoides, displasia o adelgazamiento cortical de huesos largos con o sin seudoatrosis), y antecedentes de neurofibromatosis tipo 1 en padres o hermanos.
Si bien la presentación de complicaciones es directamente proporcional al incremento de la edad,10 el pronóstico de la enfermedad generalmente es bueno, aunque acompañado de una morbilidad significativa; los tumores malignos de las vainas de los nervios periféricos por lo general tienen un pronóstico desfavorable. Las principales causas de mortalidad temprana son las neoplasias y la enfermedad vascular.
La microdeleción 17q11 se caracteriza por dismorfismo facial, retraso en el desarrollo, elevado número de neurofibromas, discapacidad intelectual y mayor riesgo de neoplasias.
La neurofibromatosis tipo 2 es un síndrome de predisposición a tumores de transmisión dominante, causado por mutaciones en el gen NF2 del cromosoma 22. Tiene una prevalencia de aproximadamente 1/60 000, y se caracteriza por la aparición de múltiples schwannomas y meningiomas que afectan ambos nervios vestibulares, lo que provoca sordera como primer síntoma; también pueden causar mareos o trastornos del equilibrio. Otros schwannomas principales afectan otros nervios craneales, espinales y periféricos; los meningiomas pueden ser intracraneales o intraespinales. Aproximadamente el 70% de estos pacientes tienen tumores de la piel. El diagnóstico se basa en estudios clínicos y por neuroimágenes; existen, además, pruebas genéticas.
El pronóstico de esta enfermedad es malo, el tratamiento es la cirugía y, ocasionalmente, la irradiación.
La neurofibromatosis tipo 3 es la forma menos común, es clínica y genéticamente distinta de los tipos 1 y 2, y se caracteriza por la presencia de múltiples schwannomas, sin afección de los nervios vestibulares. Se diagnostica generalmente en la edad adulta, afecta a menos de 1 cada 40 000 personas y se caracteriza por dolor crónico, disestesias y parestesias. Suele localizarse en la columna vertebral, los nervios periféricos y el cráneo.
La neurofibromatosis tipo 6 es una enfermedad cutánea extremadamente rara, caracterizada por la presencia de máculas de color café con leche sin otras manifestaciones de neurofibromatosis o de otra enfermedad sistémica. El tamaño de las máculas puede variar desde unos pocos milímetros hasta más de 10 cm. Su etiología es desconocida, aunque algunas formas se han asociado con mutaciones del gen NF1 (17q11.2); la transmisión es autosómica dominante y su diagnóstico se basa en la presencia de más de cinco máculas, que son benignas, no necesitan tratamiento médico y algunas pueden desaparecer con la edad.
Las complicaciones que pueden presentar los pacientes con neurofibromatosis tipo 1 son muy variadas y afectan distintos órganos de la economía, como la piel, el tejido óseo y las articulaciones; también puede manifestarse hipertensión arterial, hidrocefalia y trastornos cognitivos, entre otras. Las alteraciones vasculares son una complicación rara de esta enfermedad y suelen ser asintomáticas; pueden afectar los vasos que van desde la aorta proximal hasta las arteriolas pequeñas, incluyendo estenosis arteriales, aneurismas y malformaciones arteriovenosas.11
Entre las alteraciones vasculares descritas como complicación de la enfermedad de Von Recklinghausen se citan aneurismas y seudoaneurismas de la arteria intercostal;12-14 presentan como manifestación hemotórax, aneurisma de la arteria vertebral (la que, en un caso descrito por Uneda, presentó luego de su ruptura una fístula arteriovenosa),15 mientras que en otros casos se manifiesta por hemotórax,16,17 aneurismas de la carótida interna18 o seudoaneurisma de las ramas de la arteria subclavia.11
En lo que respecta al tratamiento, en la actualidad se utiliza la quimioterapia; sin embargo, se están desarrollando tratamientos específicos basados en estudios genómicos, con la finalidad de reemplazar o restaurar la función de la neurofibromina defectuosa.
La neurofibromatosis, fue descrita en 1882 por el patólogo alemán Friedrich Daniel von Recklinghausen, es una enfermedad autosómica dominante rara, producida por una mutación en el cromosoma 17, caracterizada por la aparición de tumores en distintos órganos.
En el 50% de los casos la mutación es heredada de unos de los padres.1
Se describen cuatro tipos de neurofibromatosis.
La neurofibromatosis tipo 1 es producida por mutaciones en el gen NF1 que inhiben la neurofibromina, considerada como un supresor tumoral2 localizado en el cromosoma 17q11.2,3-5 y en un pequeño porcentaje de casos por microdeleción 17q11.6,7 En el 50% de los casos es autosómica dominante, aunque puede producirse por mutaciones de novo en NF1; la penetrancia es del 100%.8 El diagnóstico suele ser clínico y confirmado por medio de estudios genéticos.
La neurofibromatosis tipo 2 es un síndrome de predisposición a tumores, de transmisión dominante, causado por mutaciones en el gen NF2 del cromosoma 22, y se caracteriza por la aparición de múltiples schwannomas y meningiomas que afectan a ambos nervios vestibulares provocando sordera, mareos o trastornos del equilibrio. El diagnóstico se basa en estudios clínicos y por neuroimágenes; existen pruebas genéticas. El pronóstico es malo, el tratamiento es la cirugía y ocasionalmente la irradiación.
La neurofibromatosis tipo 3 se caracteriza por la existencia de múltiples schwannomas, sin afección de los nervios vestibulares. Se diagnostica generalmente en la edad adulta, afecta a menos de 1 cada 40 000 personas y se caracteriza por dolor crónico, disestesias y parestesias.
Finalmente, la neurofibromatosis tipo 6 es una enfermedad cutánea extremadamente rara, caracterizada por la presencia de máculas de color café con leche, cuyo tamaño puede variar desde unos pocos milímetros hasta más de 10 centímetros. Su etiología es desconocida, aunque algunas formas se han asociado con mutaciones del gen NF1 (17q11.2); la transmisión es autosómica dominante y su diagnóstico se basa en la presencia de más de cinco máculas, que son benignas, no necesitan tratamiento médico y algunas pueden desaparecer con la edad.
Caso clínico
Paciente de 31 años con diagnóstico de neurofibromatosis tipo 1 y cirugía estética de maxilar inferior a los 20 años, que sufrió traumatismo facial aparentemente leve. Al tercer día de producido el trauma consultó por aumento del volumen de la mitad izquierda de la cara y el cuello, secundaria a importante hematoma con compromiso de la vía aérea, por lo que se efectuó intubación orotraqueal y se instauró ventilación mecánica invasiva. Debido a la falta de complejidad fue derivado a nuestra institución, con tubo orotraqueal y dependiente de ventilación mecánica, bajo efecto de fármacos depresores del sistema nervioso central (Ramsay: -4). Se efectuó tomografía axial computarizada (TAC) y angiotomografía, que informó presencia de masa heterogénea hiperdensa voluminosa en el tejido celular subcutáneo de la hemicara izquierda, con extensión desde la región frontal hasta la mitad del cuello, extendiéndose hasta la fosa pterigomaxilar. Extensión al espacio parafaríngeo con desplazamiento de la laringe y en parte de la tráquea. Arteria carótida permeable, flujo preferencial por carótida derecha, carótida izquierda permeable, pero fina en todo su recorrido.
Esta masa presentaba áreas de hipervascularización y estaba irrigada por una rama de la arteria subclavia y, aparentemente, de la carótida externa. Se efectuó angiografía que mostró la presencia de un seudoaneurisma dependiente de la arteria temporal (identificándosela como responsable del sangrado), observándose por medio de la arteria maxilar interna una fístula con drenaje a la vena facial superficial y pequeñas dilataciones sin aferencias factibles de embolización, por lo que se llevó a cabo dicho procedimiento cateterizándose la rama aferente del seudoaneurisma (Figura 1).
A las 48 horas se efectuó una nueva TAC que mostró presencia de masa heterogénea, correspondiente a un hematoma, desde la región superciliar izquierda hasta la totalidad del maxilar inferior, con desviación de la tráquea hacia la derecha (Figura 2). Dado el tiempo de intubación orotraqueal (10 días) y la imposibilidad de retiro del tubo orotraqueal por inminencia de obstrucción de la vía aérea, se efectuó traqueostomía (Figura 3), con lo que el paciente fue desvinculado del ventilador sin complicaciones. Se drenaron a los 14 días del evento mediante punción guiada por ecografía, aproximadamente 350 centímetros cúbicos (Figura 4). El paciente evolucionó favorablemente, con buena mecánica ventilatoria, con ingesta de alimentos con buena tolerancia.
Discusión
La neurofibromatosis tipo 1 se produce por mutaciones en el gen NF1, lo que inhibe la neurofibromina, considerada como un supresor tumoral localizado, en el cromosoma 17q11.2 y en un pequeño porcentaje de casos por microdelección 17q11. Es autosómica dominante, aunque en el 50% de los casos se produce por mutaciones de novo en NF1; la penetrancia es del 100%. Su prevalencia aproximadamente es de 1/30003 nacidos vivos y afecta por igual a ambos sexos. Suele manifestarse en la infancia, aunque los signos pueden estar presentes desde el nacimiento, y se manifiesta por cambios en la coloración de la piel (máculas de color café con leche), pecas intertriginosas que aparecen a partir de los 5 año y, posteriormente, formación de múltiples neurofibromas cutáneos y subcutáneos que, con el transcurso de los años, se incrementan en número y tamaño. Los neurofibromas que crecen a lo largo del nervio pueden causar deformaciones, dolor o trastornos funcionales, y pueden malignizarse. Las manifestaciones oculares son los gliomas de la vía óptica y los hamartomas del iris (nódulos de Lisch). Puede presentarse osteopenia, osteoporosis, sobrecrecimiento óseo, macrocefalia, escoliosis, displasia esquelética y seudoartrosis; también puede aparecer hipertensión arterial, alteraciones vasculares, tumores intracraneales, tumores malignos de las vainas de los nervios periféricos y, ocasionalmente, convulsiones e hidrocefalia; son frecuentes los déficits cognitivos y las dificultades del aprendizaje.
El diagnóstico se efectúa con la presencia de dos o más de los siguientes signos:9 más de seis máculas de color café con leche de 5 mm en los pacientes prepúberes, y mayores de 15 mm en los pospúberes; dos o más neurofibromas de cualquier tipo o un neurofibroma plexiforme; glioma del nervio óptico; signo de Crowe: efélides axilares o inguinales; dos o más nódulos de Lisch (hamartomas del iris); displasias óseas específicas (displasia del esfenoides, displasia o adelgazamiento cortical de huesos largos con o sin seudoatrosis), y antecedentes de neurofibromatosis tipo 1 en padres o hermanos.
Si bien la presentación de complicaciones es directamente proporcional al incremento de la edad,10 el pronóstico de la enfermedad generalmente es bueno, aunque acompañado de una morbilidad significativa; los tumores malignos de las vainas de los nervios periféricos por lo general tienen un pronóstico desfavorable. Las principales causas de mortalidad temprana son las neoplasias y la enfermedad vascular.
La microdeleción 17q11 se caracteriza por dismorfismo facial, retraso en el desarrollo, elevado número de neurofibromas, discapacidad intelectual y mayor riesgo de neoplasias.
La neurofibromatosis tipo 2 es un síndrome de predisposición a tumores de transmisión dominante, causado por mutaciones en el gen NF2 del cromosoma 22. Tiene una prevalencia de aproximadamente 1/60 000, y se caracteriza por la aparición de múltiples schwannomas y meningiomas que afectan ambos nervios vestibulares, lo que provoca sordera como primer síntoma; también pueden causar mareos o trastornos del equilibrio. Otros schwannomas principales afectan otros nervios craneales, espinales y periféricos; los meningiomas pueden ser intracraneales o intraespinales. Aproximadamente el 70% de estos pacientes tienen tumores de la piel. El diagnóstico se basa en estudios clínicos y por neuroimágenes; existen, además, pruebas genéticas.
El pronóstico de esta enfermedad es malo, el tratamiento es la cirugía y, ocasionalmente, la irradiación.
La neurofibromatosis tipo 3 es la forma menos común, es clínica y genéticamente distinta de los tipos 1 y 2, y se caracteriza por la presencia de múltiples schwannomas, sin afección de los nervios vestibulares. Se diagnostica generalmente en la edad adulta, afecta a menos de 1 cada 40 000 personas y se caracteriza por dolor crónico, disestesias y parestesias. Suele localizarse en la columna vertebral, los nervios periféricos y el cráneo.
La neurofibromatosis tipo 6 es una enfermedad cutánea extremadamente rara, caracterizada por la presencia de máculas de color café con leche sin otras manifestaciones de neurofibromatosis o de otra enfermedad sistémica. El tamaño de las máculas puede variar desde unos pocos milímetros hasta más de 10 cm. Su etiología es desconocida, aunque algunas formas se han asociado con mutaciones del gen NF1 (17q11.2); la transmisión es autosómica dominante y su diagnóstico se basa en la presencia de más de cinco máculas, que son benignas, no necesitan tratamiento médico y algunas pueden desaparecer con la edad.
Las complicaciones que pueden presentar los pacientes con neurofibromatosis tipo 1 son muy variadas y afectan distintos órganos de la economía, como la piel, el tejido óseo y las articulaciones; también puede manifestarse hipertensión arterial, hidrocefalia y trastornos cognitivos, entre otras. Las alteraciones vasculares son una complicación rara de esta enfermedad y suelen ser asintomáticas; pueden afectar los vasos que van desde la aorta proximal hasta las arteriolas pequeñas, incluyendo estenosis arteriales, aneurismas y malformaciones arteriovenosas.11
Entre las alteraciones vasculares descritas como complicación de la enfermedad de Von Recklinghausen se citan aneurismas y seudoaneurismas de la arteria intercostal;12-14 presentan como manifestación hemotórax, aneurisma de la arteria vertebral (la que, en un caso descrito por Uneda, presentó luego de su ruptura una fístula arteriovenosa),15 mientras que en otros casos se manifiesta por hemotórax,16,17 aneurismas de la carótida interna18 o seudoaneurisma de las ramas de la arteria subclavia.11
En lo que respecta al tratamiento, en la actualidad se utiliza la quimioterapia; sin embargo, se están desarrollando tratamientos específicos basados en estudios genómicos, con la finalidad de reemplazar o restaurar la función de la neurofibromina defectuosa.
Fernando Ricard Racca Velásquez, Munro, Argentina,
e-mail: fracca@intramed.net
1. Huson SM. Neurofibromatosis: historical perspective, classification and diagnostic criteria. The neurofibromatoses: A pathogenic and clinical overview. Edited by SM Huson, RAC Hughes; Chapman & Hall Medical; 1994. Pp. 1-22.
2. Valero MC, Martin Y, Hernandez-Imaz E, Hrnández A, Meleán G, Valero A, et al. A highly sensitive genetic protocol to detect NF1 mutations. J Mol Diagn 13(2):113-122, 2011.
3. Rasmussen SA, Friedman JM. NF1 gene and neurofibromatosis 1. Am J Epidemiol 151:33-40, 2000.
4. Cawthon R, Weiss R, Xu G, Viskochil D, Culver M, Stevens J, et al. Major segment of the neurofibromatosis type 1 gene: cDNA sequence, genomic structure and point mutations. Cell 62:193-201, 1990.
5. Wallace M, Marchuk D, Andersen L, Letcher R, Odeh H, Saulino A, et al. Type 1 neurofibromatosis gene:identification of a large transcript disrupted in three NF1 patients. Science 249:181-186, 1990.
6. Castle B, Baser ME, Huson SM, Cooper DN, Upadhyaya M. Evaluation of genotype-phenotype correlations in neurofibromatosis type 1. J Med Genet 40:e109, 2003.
7. Pasmant E, Sabbagh A, Spurlock G, Laurendeau I, Grillo E, Hamel M, et al. NF1 microdeletions in neurofibromatosis type 1: from genotype to phenotype. Hum Mutat 31:E1506-1518, 2010.
8. Jouhilahti EM, Peltonen S, Heape AM. Peltonen J. The pathoetiology of neurofibromatosis 1. Am J Pathol 178 (5):1932-1939, 2011.
9. NIH Consensus Development Conference. Neurofibromatosis Conference Statement. Arch Neurol 45:475-578, 1988.
10. Jett K, Friedman JM. Clinical and genetic aspects of neurofibromatosis 1. Genet Med 12:1-11, 2010.
11. Negreira KE, Lichtenberger JP, Allais B, Alhaddad A, Bernetich M, Jain V. Subclavian artery branch pseudoaneurysm rupture with massive hemothorax ina patient with neurofibromatosis type 1. Chest 157(4):103-105, 2020.
12. Chang WC, Hsu HH, Chang H, Chen CY. Spontaneous hemothorax caused by a ruptured intercostals artery aneurysm in von Recklinghausen's neurofibromatosis. J Formos Med Assoc 104:286-289, 2005.
13. Miura T, Kawano Y, Chujo M, Miyawaki M, Mori H, Kawahara K. Spontaneous hemothorax in patients with von Recklinghausen's disease. Jpn J Thorac Cardiovasc Surg 53(12):649-652, 2005.
14. Imamura F, Mase K, Marumori T, Kamiga M. Hemothorax associated with von Recklinghausen's disease; reportof a case. Kyobu Geka 60(7):599-601, 2007.
15. Uneda A, Suzuki K, Okubo S, Hirashita K, Yunoki M, Yoshino K. Neurofibromatosis type 1 - Associated extracranial vertebral artery aneurysm complicated by vertebral arteriovenous fistula after rupture: case report and literature review. World Neurosurg 96:13-18, 2016.
16. Han KS, Lee KM, Kim BJ, Kwun BD, Choi SK, Lee SH. Life-threatening hemothorax caused by spontaneous extracranial vertebral aneurysm rupture in neurofibromatosis type 1. World Neurosurg 130:157-159, 2019.
17. Bidad R, Hall C, Blohm E. Fatal tension cases. Emerg Med 18:3(4):364-368, 2019.
18. Onkendi E, Moghaddam MB, Oderich GS. Internal carotid artery aneurysms in a patient with neurofibromatosis type 1. Vasc Endovascular Surg 44(6):511-514, 2010.
Artículos publicados por el autor
(selección)
Racca Velásquez, Fernando Paquimeningitis hipertrofica pos infecciosa. Presentación de caso clinico Salud i Ciencia 23(23):343-347, 2019
Racca Velasquez, Fernando 20. %u201CHIPERACTIVIDAD SIMPATICA PAROXISTICA: PRESENTACION DE UN CASO%u201D. Salud i Ciencia 22(22):652-655, 2017
Racca Velásquez Fernando %u201CSINDROME DE GUILLAIN BARRÉ: REPORTE DE UN CASO DE PRESENTACIÓN ATÍPICA%u201D. Revista de Medicina Interna 12(12):184-190, 2016
Racca Velásquez Fernando SINDROME DE GUILLAIN BARRÉ: REPORTE DE UN CASO DE PRESENTACIÓN ATÍPICA%u201D. Revista de Medicina Interna 12(12):184-190, 2016
Racca Velasquez Fernando %u201CHIPERTENSION PULMONAR ASOCIADA CON LUPUS ERITEMATOSO SISTEMICO%u201D. Salud i Ciencia 22(22):259-263, 2016
Racca Velasquez Fernando HIPERTENSION PULMONAR ASOCIADA CON LUPUS ERITEMATOSO SISTEMICO%u201D. Salud i Ciencia 22(22):259-263, 2016
Racca velasquez Fernando Validacion del Modelo matemática predictivo de Mortalidad para la Hemorragia Supratentorial espontanea Salud i Ciencia 21(21):598-603, 2015
Racca Velásquez Fernando Catatonia letal de Stauder Revista de medicina Interna 11(11):57-60, 2015
Racca Velásquez fernando HEMORRAGIA SUPRATENTORIAL ESPONTANEA: Un modelo matemático predictivo de mortalidad Salud i Ciencia 20(20):368-372, 2014
Racca Velásquez Fernando HEMORRAGIA SUPRATENTORIAL ESPONTANEA: Edad y Sexo como predictores de mortalidad Revista de medicina Interna 9(9):122-125, 2014
Racca Velasquez Fernando INCIDENCIA DE RUPTURA ALVEOLAR INDUCIDA POR LA VENTILACION MECANICA REVISTA DE MEDICINA INTERNA 7(7):173-178, 2011
Racca Velásquez Fernando INCIDENCIA DE BACTERIEMIA POR CATETER Revista de medicina Interna 6(6):129-131, 2010
Racca Velásquez, Fernando Paquimeningitis hipertrofica pos infecciosa. Presentación de caso clinico Salud i Ciencia 23(23):343-347, 2019
Racca Velasquez, Fernando 20. %u201CHIPERACTIVIDAD SIMPATICA PAROXISTICA: PRESENTACION DE UN CASO%u201D. Salud i Ciencia 22(22):652-655, 2017
Racca Velásquez Fernando %u201CSINDROME DE GUILLAIN BARRÉ: REPORTE DE UN CASO DE PRESENTACIÓN ATÍPICA%u201D. Revista de Medicina Interna 12(12):184-190, 2016
Racca Velásquez Fernando SINDROME DE GUILLAIN BARRÉ: REPORTE DE UN CASO DE PRESENTACIÓN ATÍPICA%u201D. Revista de Medicina Interna 12(12):184-190, 2016
Racca Velasquez Fernando %u201CHIPERTENSION PULMONAR ASOCIADA CON LUPUS ERITEMATOSO SISTEMICO%u201D. Salud i Ciencia 22(22):259-263, 2016
Racca Velasquez Fernando HIPERTENSION PULMONAR ASOCIADA CON LUPUS ERITEMATOSO SISTEMICO%u201D. Salud i Ciencia 22(22):259-263, 2016
Racca velasquez Fernando Validacion del Modelo matemática predictivo de Mortalidad para la Hemorragia Supratentorial espontanea Salud i Ciencia 21(21):598-603, 2015
Racca Velásquez Fernando Catatonia letal de Stauder Revista de medicina Interna 11(11):57-60, 2015
Racca Velásquez fernando HEMORRAGIA SUPRATENTORIAL ESPONTANEA: Un modelo matemático predictivo de mortalidad Salud i Ciencia 20(20):368-372, 2014
Racca Velásquez Fernando HEMORRAGIA SUPRATENTORIAL ESPONTANEA: Edad y Sexo como predictores de mortalidad Revista de medicina Interna 9(9):122-125, 2014
Racca Velasquez Fernando INCIDENCIA DE RUPTURA ALVEOLAR INDUCIDA POR LA VENTILACION MECANICA REVISTA DE MEDICINA INTERNA 7(7):173-178, 2011
Racca Velásquez Fernando INCIDENCIA DE BACTERIEMIA POR CATETER Revista de medicina Interna 6(6):129-131, 2010
Está expresamente prohibida la redistribución y la redifusión de todo o parte de los
contenidos de la Sociedad Iberoamericana de Información Científica (SIIC) S.A. sin
previo y expreso consentimiento de SIIC.
ua91218

