SINDROME DE OVARIO POLIQUISTICO COMO MODELO CLINICO DE EXPOSICION PRENATAL A ANDROGENOS
(especial para SIIC © Derechos reservados)
Coautores
Manuel Alejandro Maliqueo Yevilao* Pedro Pablo Rojas García** Sergio Edmundo Recabarren Morgado***
Profesor Asistente. Facultad de Medicina. Universidad de Chile*
Profesor Asistente. Facultad de Medicina Veterinaria, Universidad de Concepción**
Profesor Titular. Facultad de Medicina Veterinaria, Universidad de Concepción***
Recepción del artículo: 30 de agosto, 2004
Aprobación: 30 de septiembre, 2004
 Conclusión breve
Recientemente, hemos podido establecer que las embarazadas con síndrome de ovario poliquístico presentan niveles androgénicos significativamente más altos que las embarazadas normales, lo que abre la posibilidad que los hijos de estas mujeres pudiesen haber estado sometidos a un ambiente esteroidal anormal durante su vida fetal.
Resumen
Se ha propuesto que la exposición prenatal a andrógenos (EPA) durante la vida fetal en forma experimental, accidental o patológica podría producir una serie de cambios del eje reproductivo y de la homeostasis glucídica del feto femenino que se harían evidentes en su vida posnatal y semejarían aquellos descritos en el síndrome de ovario poliquístico (SOP). Si bien la EPA podría estar involucrada en el desarrollo del SOP no se ha establecido si la madre con SOP podría constituir una fuente de exceso de andrógenos para el feto. En la hiperplasia virilizante congénita, un modelo clásico de EPA, podría ser la corteza suprarrenal hiperandrogénica del propio feto el origen del exceso prenatal de andrógenos, mientras que en el SOP este origen parece más incierto. Recientemente, hemos podido establecer que las embarazadas con SOP presentan niveles androgénicos significativamente más altos que las embarazadas normales, lo que abre la posibilidad que los hijos de estas mujeres pudiesen haber estado sometidos a un ambiente esteroidal anormal durante su vida fetal, el cual podría afectar tanto al feto femenino como masculino. Futuros estudios permitirán evaluar el efecto a largo plazo de este exceso de andrógenos prenatales sobre la descendencia de madres con SOP con el fin de establecer si el SOP debería ser tratado durante el embarazo con el fin de evitar el exceso de andrógenos.
Palabras clave
Síndrome de ovario poliquístico, exposición prenatal a andrógenos, androgenización fetal
Clasificación en siicsalud
Conclusión breve
Recientemente, hemos podido establecer que las embarazadas con síndrome de ovario poliquístico presentan niveles androgénicos significativamente más altos que las embarazadas normales, lo que abre la posibilidad que los hijos de estas mujeres pudiesen haber estado sometidos a un ambiente esteroidal anormal durante su vida fetal.
Resumen
Se ha propuesto que la exposición prenatal a andrógenos (EPA) durante la vida fetal en forma experimental, accidental o patológica podría producir una serie de cambios del eje reproductivo y de la homeostasis glucídica del feto femenino que se harían evidentes en su vida posnatal y semejarían aquellos descritos en el síndrome de ovario poliquístico (SOP). Si bien la EPA podría estar involucrada en el desarrollo del SOP no se ha establecido si la madre con SOP podría constituir una fuente de exceso de andrógenos para el feto. En la hiperplasia virilizante congénita, un modelo clásico de EPA, podría ser la corteza suprarrenal hiperandrogénica del propio feto el origen del exceso prenatal de andrógenos, mientras que en el SOP este origen parece más incierto. Recientemente, hemos podido establecer que las embarazadas con SOP presentan niveles androgénicos significativamente más altos que las embarazadas normales, lo que abre la posibilidad que los hijos de estas mujeres pudiesen haber estado sometidos a un ambiente esteroidal anormal durante su vida fetal, el cual podría afectar tanto al feto femenino como masculino. Futuros estudios permitirán evaluar el efecto a largo plazo de este exceso de andrógenos prenatales sobre la descendencia de madres con SOP con el fin de establecer si el SOP debería ser tratado durante el embarazo con el fin de evitar el exceso de andrógenos.
Palabras clave
Síndrome de ovario poliquístico, exposición prenatal a andrógenos, androgenización fetal
Clasificación en siicsalud
Artículos originales> Expertos del Mundo>
página www.siicsalud.com/des/expertos.php/69977
Especialidades
 Principal: Farmacología, Obstetricia y Ginecología,
Principal: Farmacología, Obstetricia y Ginecología,
Relacionadas: Bioquímica, Diagnóstico por Laboratorio, Endocrinología y Metabolismo, Medicina Interna,
Enviar correspondencia a:
Sir Petermann, Teresa
Patrocinio y reconocimiento
Agradecimientos: Esta línea de investigación está financiada por el Fondo de Desarrollo Científico y Tecnológico de Chile (FONDECYT) mediante los proyectos 1030487, 1020232 y 1970291.
POLYCYSTIC OVARIC SYNDROME AS CLINICAL MODEL OF ANDROGENS PRENATAL EXPOSURE
Abstract
It has been proposed that androgen excess during fetal life either experimentally, accidentally or pathologically will produce changes in the reproductive axis and glucose homeostasis of the adult female which resemble those observed in the polycystic ovary syndrome (PCOS). Although androgen excess during pregnancy has been proposed to be involved in the development of PCOS, it has not been established if PCOS mothers could provide a potential source of androgen excess for the fetus. In women with a classical 21-hydroxylase deficiency, the fetal suprarrenal cortex of the fetus will be the source of the androgen excess. In the case of the PCOS, this origin is uncertain. We have recently demonstrated a significant increase in serum androgen concentrations during pregnancy in PCOS women compared to normal pregnant women, which opens the possibility that children of PCOS women could be exposed to an abnormal steroid milieu during fetal life affecting either the female or male fetus. Further studies are needed to evaluate the potential long effect of this prenatal androgen excess on baby girls and boys born to PCOS mother in order to establish if PCOS women should be treated during pregnancy in order to avoid the androgen excess.
Key words
Polycystic ovary syndrome, prenatal androgen excess, fetal androgen excess
SINDROME DE OVARIO POLIQUISTICO COMO MODELO CLINICO DE EXPOSICION PRENATAL A ANDROGENOS
(especial para SIIC © Derechos reservados)
Artículo completo
El SOP: un síndrome de etiología no resuelta
El síndrome de ovario poliquístico (SOP), también denominado hiperandrogenismo ovárico funcional o anovulación crónica hiperandrogénica, es la causa más común de hiperandrogenismo, con una incidencia de un 3% tanto en mujeres adolescentes como adultas. Se estima además que está presente en 75% de las mujeres hirsutas y en 10% de las mujeres premenopaúsicas. Su etiología es incierta y se encuentra en estrecha asociación con la resistencia insulínica (RI) y la diabetes tipo 2, las que suelen presentarse precozmente.1-2 En los últimos años se puso de manifiesto, además, que este trastorno no solo está limitado a la mujer en etapa reproductiva sino que puede manifestarse desde el período prepuberal y posiblemente aun antes.3-5
El SOP representa una combinación de anovulación, hiperandrogenismo e hiperinsulinemia, por lo general acompañados de hipersecreción de LH y la presencia de ovarios poliquísticos en la ultrasonografía. Las manifestaciones clínicas del SOP son heterogéneas y varían de acuerdo con la edad de la paciente. Por lo general, se inician en el período perimenárquico con la aparición de alteraciones menstruales entre las que destacan la oligomenorrea,6 manifestaciones de hiperandrogenismo y obesidad androide.
Durante este período es particularmente difícil sospechar clínicamente el SOP debido a los cambios fisiológicos del eje somatotrópico y reproductivo que presentan las niñas durante esta etapa de su desarrollo sexual.7 Es importante destacar, además, que estudios recientes establecen que el SOP puede aparecer antes de este período con adrenarquia prematura, pubertad adelantada e hiperinsulinismo, los que se relacionan con retraso del crecimiento intrauterino y el nacimiento de un niño pequeño para la edad gestacional (PEG).3,8
Debido a la heterogeneidad de sus manifestaciones clínicas, su definición es difícil de establecer. En los últimos años se propusieron dos definiciones: presencia de hiperandrogenismo asociado con anovulación crónica sin otra causa específica de enfermedad suprarrenal o hipofisiaria;9 y disfunción ovárica caracterizada por hiperandrogenismo y morfología de ovario poliquístico.10
La complejidad de la fisiopatología del SOP es de tal magnitud que su etiología permanece incierta. En ella destacan tres tipos de alteraciones interrelacionadas entre sí: una disfunción neuroendocrina caracterizada por hipersecreción de LH, un trastorno metabólico caracterizado por resistencia a la insulina e hiperinsulinemia y disfunción de la esteroidogénesis ovárica y suprarrenal. Esta última es estrictamente necesaria para que se constituya el síndrome y consiste en la hiperactividad de la enzima CYP17 (citocromo P450), la cual tiene actividad 17-hidroxilasa y 17-20 liasa y cataliza el paso de progesterona a 17αOH progesterona (17OHP), y de ésta a androstenediona (Δ4A). Se ha sugerido que esta disfunción podría ser exclusiva del SOP, y que podría ser un evento primario o secundario al exceso de LH, de insulina o de ambas.11-12 El aumento de la LH o de la insulina en el SOP lleva a una sobreexpresión de la enzima, que conduce a mayor producción de andrógenos intraováricos y anovulación crónica por inhibición del desarrollo folicular. Recientemente se determinó el gen para esta enzima y se propusó una posible disfunción relacionada con el SOP.13
Además, aproximadamente 25% de las pacientes con SOP presentan exceso de andrógenos suprarrenales, expresado por aumento moderado de DHEAS e hiperrespuesta a ACTH.14 Este fenómeno parece estar genéticamente determinado y vinculado probablemente a una actividad exagerada de la CYP1715-16 y, menos probablemente, a otras disfunciones enzimáticas. En todo caso, en el SOP podría existir una interrelación fisiopatológica entre el ovario y la suprarrenal, al actuar los andrógenos suprarrenales como precursores de andrógenos ováricos y viceversa.17-18
La disfunción neuroendocrina, caracterizada por aumento de la secreción de LH y disminución de la secreción de FSH fue una de las primeras características descritas en el SOP. Aún no se establece si es una disfunción hipotalámica primaria o secundaria a los niveles elevados de insulina19 o al retrocontrol de hormonas ováricas.20-22 Se pudo establecer que la secreción ultradiana y circadiana de LH está alterada en mujeres con SOP,23-25 sin que se hayan identificado alteraciones específicas de algunos neurotransmisores que la expliquen, lo que llevó a plantear que en ciertos casos de SOP el aumento de la secreción de LH podría ser expresión de una disfunción hipotalámica, la que se debería a una “reprogramación” del generador de pulsos de GnRH por una exposición prenatal o prepuberal a andrógenos.26 En adolescentes con SOP también se documentó alteración de la secreción circadiana de LH,27 pulsatilidad aumentada de LH, cociente LH/FSH elevado y aumento de andrógenos ováricos compatible con un incremento en la actividad de 17 hidroxilasa.28-29 Estas alteraciones en la secreción de gonadotropinas pueden manifestarse en forma longitudinal durante la pubertad de niñas con anovulación,29 en especial en niñas con pubarquia prematura;30 sin embargo, no se encontraron anormalidades intrínsecas del eje somatotrófico (GH-IGF-1, IGF-BP3) en estas pacientes.8
Por otro lado se demostró que la disminución del peso corporal,31 la resección cuneiforme y fulguración del ovario y el uso de drogas que disminuyen la hiperinsulinemia,32 restablecen transitoriamente la ciclicidad ovárica, lo que sugiere que en algunos casos las alteraciones de la secreción gonadotrópica serían secundarias a la disfunción metabólica o del retrocontrol anormal de las hormonas ováricas, en el cual los andrógenos podrían jugar un papel preponderante.22,33-34
Respecto de la disfunción metabólica del SOP, la mayoría de estas pacientes presentan hiperinsulinemia, la que podría ser considerada marcador de resistencia periférica a la insulina. La sensibilidad tisular a la insulina, medida por diferentes métodos, está reducida significativamente en el SOP, tanto en mujeres obesas como no obesos, en comparación con controles sanos de igual índice de masa corporal.35-37 La asociación entre hiperinsulinemia y aumento de la biosíntesis de andrógenos en mujeres con SOP es apoyada por numerosos estudios in vivo e in vitro,11,38-40 lo que llevó a proponer que podría ser la RI la que al condicionar hiperinsulinemia, llevaría a hiperandrogenemia a través del efecto estimulador de la insulina sobre las células tecales del ovario y las reticulares de la suprarrenal. No obstante, no se puede descartar totalmente el efecto de los andrógenos sobre la génesis de la hiperinsulinemia. Un estudio reciente demuestra que los andrógenos podrían tener efecto directo sobre la función de los islotes pancreáticos favoreciendo la expresión del gen de insulina y la secreción de insulina.41 En todo caso, el mecanismo por el cual se genera RI en el SOP no está claro. Se propuso que en estas pacientes habría una alteración de los eventos posreceptor.42-45 En el SOP, semejante a lo descrito en la diabetes tipo 2, la RI parece preceder a la disminución de la tolerancia a la glucosa. No todas las pacientes con SOP con RI desarrollan intolerancia a la glucosa y diabetes 2, por lo que se propuso que debe coexistir una disfunción betapancreática, la cual podría ser condicionada por el mismo defecto que genera la RI, o como por un imprinting prenatal patológico por efecto de los andrógenos sobre los islotes pancreáticos.
En consecuencia, la etiología del SOP aún no está esclarecida y podría incluir una combinación de factores genéticos y ambientales. Sin embargo, no se ha podido identificar una etiología única, que tenga un poder predictivo en el desarrollo del SOP.
Ambiente intrauterino, un posible mecanismo etiológico en el desarrollo del SOP
Los primeros indicios que vincularon un posible papel de una exposición excesiva de andrógenos en la vida fetal con el desarrollo ulterior de SOP en la vida posnatal derivaron de observaciones en mujeres portadoras de un déficit clásico de la 21-hidroxilasa (figura 1) que fueron adecuadamente tratadas y que por ende presentaron normalización de sus andrógenos. Durante su vida adulta, las pacientes desarrollaron rasgos típicos de SOP como anovulación, hipersecreción de LH, RI y aspecto poliquístico de los ovarios. Lo anterior llevó a proponer que el exceso de andrógenos durante la vida fetal –ya sea de origen ovárico o suprarrenal– podría constituir una alteración hormonal que sería necesaria para el desarrollo de SOP en la vida posnatal.46 No obstante, los estudios prospectivos en humanos fueron limitados, lo que dio origen a la utilización de modelos animales.
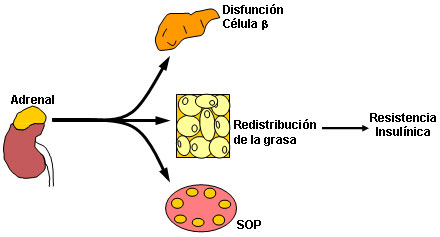
Figura 1. En humanos un modelo clásico de EPA es la hiperplasia virilizante congénita por déficit de la 21-hidroxilasa, una enzima clave en la esteroidogénesis suprarrenal. En ella, la excesiva producción de andrógenos por la suprarrenal del feto produce autoandrogenización, lo que lleva a disfunción betapancreática, redistribución de la grasa y cambios de tipo poliquístico en la morfología ovárica.
Estudios en monas Rhesus y hembras ovinas con exposición prenatal a andrógenos (EPA) demuestran que estas hembras desarrollan características endocrinológicas y anatómicas de los ovarios que típicamente se asemejan al SOP de los humanos, como hiperandrogenismo ovárico y suprarrenal, hipersecreción de LH, anovulación asociada a hiperinsulinemia y ovarios aumentados de volumen de tipo multifolicular.26,47-53 Estos resultados sugieren que una discreta exposición prenatal a andrógenos podría explicar mucho de los rasgos típicos del SOP. En este sentido cobran gran interés las primeras observaciones efectuadas por nuestro grupo en mujeres con SOP en amenorrea de la lactancia, las cuales presentaban ovarios aumentados de volumen y mayor concentración de androstenediona que las nodrizas normales.25 (tabla 1). Estos resultados nos permitieron especular que los ovarios de estas mujeres permanecieron activados durante el embarazo manteniendo una secreción androgénica elevada, lo que eventualmente podría haber androgenizado un feto femenino.
Tabla 1Posteriormente, logramos demostrar que efectivamente las embarazadas con SOP presentaban concentraciones de testosterona y androstenediona significativamente más altas que las embarazadas normales (figura 2) lo que apoyó nuestra teoría de que el SOP podría conducir a EPA.54 De esta observación deriva justamente la importancia de evaluar los posibles efectos en la descendencia de madres con SOP. Los niños y niñas de estas mujeres nacieron con sus genitales normales. No obstante, como se demostró en el modelo de androgenización prenatal en hembras ovinas y en monos,26,48,55 no se requiere una exposición excesivamente alta a andrógenos para producir cambios metabólicos y hormonales. Otro aspecto apasionante de la EPA es su posible relación con el desarrollo de RI, hiperinsulinemia y falla betapancréatica, que la relacionan estrechamente al SOP humano. Un estudio reciente en monas Rhesus androgenizadas por EPA establece que la exposición prenatal a andrógenos altera la sensibilidad a la insulina y la función betapancreática.49

Figura 2. Concentraciones de androstenediona y testosterona en embarazadas normales (EN) y embarazadas con SOP (ESOP) durante las semanas 10 a 16 y 22 a 28 de gestación. Los valores se expresan en mediana ± SEM. *p < 0.05 ajustado por índice de masa corporal (adaptado de Sir-Petermann y col. Hum Reprod. 2002; 17:2573-2579).
De lo anteriormente expuesto se desprende que la EPA puede afectar la función reproductiva de la hembra o de la mujer. No obstante, un ambiente intrauterino anormal también podría afectar al feto masculino de una madre con SOP a través de varios mecanismos que a pesar de no haber sido formalmente descritos en la literatura se pueden inferir de otras observaciones. Si bien es difícil especular que un ambiente hiperandrogénico pudiera afectar la función neuroendocrina reproductiva futura del feto masculino de una madre con SOP, un exceso de andrógenos puede: a) constituir el sustrato para una mayor síntesis de estrógenos por la placenta, modificando el ambiente esteroidal en que el feto masculino se encuentra inmerso; b) constituir el sustrato para una mayor síntesis de estrógenos en el testículo, modificando el microambiente testicular durante el período fetal; c) modificar la función neuroendocrina reproductiva por un efecto directo de los andrógenos o por su conversión a estrógenos, y d) alterar la sensibilidad a la insulina y la función betapancreática.
Además de lo anteriormente expuesto, debemos recordar que tanto el feto femenino como el masculino de una madre con SOP se desarrollarán en un ambiente materno hiperinsulinémico,54 lo cual también podría influir en la concentración de esteroides. A este respecto se demostró en citotrofoblasto humano que la insulina inhibe la actividad de la aromatasa y estimula la actividad de la 3β hidroxiesteroide deshidrogenasa, lo cual favorecería una mayor concentración de andrógenos.56-57
Por lo tanto, sobre la base de las observaciones clínicas y experimentales queda establecido que la EPA podría jugar un papel etiopatogénico en el desarrollo del SOP. No obstante, lo que está menos explorado es si la madre con SOP constituye un modelo clínico de EPA para su descendencia (figura 3). En el modelo clínico clásico de EPA, como es la hiperplasia virilizante congénita, el origen del exceso prenatal de andrógenos podría ser la corteza suprarrenal hiperandrogénica del propio feto, mientras que en el SOP este origen es más incierto pudiendo ser la gónada fetal la que se activa generando autoandrogenización, como se observa en la hiperplasia virilizante congénita y fue propuesto recientemente por Abbott y col.58 o es la madre con SOP la que traspasa un exceso de andrógenos al feto, semejante a lo que ocurre en el modelo experimental.54 En esta última eventualidad cabe preguntarse si estas mujeres deberían ser tratadas durante el embarazo considerando que son más hiperandrogénicas e hiperinsulinémicas que las embarazadas controles.
Figura 3. Origen del síndrome de ovario poliquístico: ¿una condición prenatal
Recientemente, otro grupo de investigadores59 enfatiza el ambiente intrauterino adverso de la madre con SOP y concluye que el uso de metformina durante el embarazo disminuye el desarrollo de diabetes gestacional y previene el exceso de andrógenos para el feto. No obstante, antes de sugerir un manejo farmacológico de la embarazada con SOP, nosotros proponemos en primer lugar que la mujer con SOP debería lograr el embarazo en las mejores condiciones metabólicas y de peso corporal. Con lo cual evidentemente podría disminuir el riesgo de diabetes gestacional y de síndrome hipertensivo del embarazo. En segundo lugar, deberían establecerse las repercusiones reales de este ambiente intrauterino adverso sobre la descendencia de mujeres con SOP antes de proponer un manejo farmacológico de ellas durante el embarazo. Actualmente estamos evaluando en mayor profundidad el impacto de la exposición prenatal a andrógenos sobre la función metabólica y reproductiva masculina y femenina, para lo cual estamos utilizando un modelo experimental de EPA constituido por borregas y corderos nacidos de hembras ovinas androgenizadas y un modelo clínico formado por hijos e hijas de mujeres con síndrome de ovario poliquístico.
Los autores no manifiestan conflictos.
Bibliografía del artículo
- Ehrmann DA, Barnes RB, Rosenfield EL y col. Prevalence of impaired glucose tolerance and diabetes in women with polycystic ovary syndrome. Diabetes Care 1999; 22:141-146.
- Legro RS, Kunselman A, Dodson W y col. Prevalence and predictors risk for type 2 diabetes mellitus and impaired glucose tolerance in polycystic ovary syndrome: a prospective, controlled study in 254 affected wome. J. Clin. Endocrinol. Metab. 1999; 84:165-169.
- Ibáñez L, Potau N, Francois I y col. Precocious pubarche, hyperinsulinism and ovarian hyperandrogenism in girls: relation to reduced fetal growth. J. Clin. Endocrinol. Metab. 1998; 83:3558-3562.
- Ibáñez L, Potau N, Chacon P y col. Hyperinsulinemia, dyslipidemia and cardiovascular risk in girls with a history of premature pubarche. Diabetologia 1998; 41:1057-1063.
- Ibáñez L, DiMartino-Nardi J, Potau N y col. Premature adrenarche-normal variant forerunner of adult disease. Endrocr Rev 2000; 21:671-696.
- Van Hooff MHA, Voorhozst FJ, Kaptein MBM y col. Endocrine features of polycystic ovary syndrome in a random population sample of 14-16 year old adolescents. Hum Reprod 1999; 14:2223-2229.
- Rosenfield RL, Ghai K, Erhmann DA y col. Diagnosis of the polycystic ovary syndrome in adolescence: comparision of adolescent and adult hyperandrogenism. J. Pediatr. Endocrinol. Metab. 2000; 13(Supp 5):1285-1289.
- Apter D, Bützow T, Laughlin GA y col. Metabolic features of polycystic ovary syndrome are found in adolescent girls with hyperandrogenism. J. Clin. Endocrinol. Metab. 1995; 80:2966-2973.
- Zawdaki JK, Dunaif A. Diagnostic criteria for polycystic ovary syndrome: towards a rationale approach. En: Polycystic ovary syndrome, ed. por Dunaif A, Givens JR, Haseltine F, Merriam GR. Boston: Blackwell 1992; 377-384.
- Rotterdam ESHRE/ASRM-Sponsored PCOS consensus workshop group. Revised 2003 consensus on diagnostic criteria and long-term health risks related to polycystic ovary syndrome (PCOS). Hum Reprod 2004; 19:41-47.
- Bergh C, Carlsson B, Olsson JH. Regulation of androgen production in cultured human thecal cells by insulin-like growth factor I and insulin. Fertil. Steril. 1993; 59:323-331.
- Ehrmann DA, Barnes RB, Rosenfield EL. Polycystic ovary syndrome as a form functional ovarian hyperandrogenism due to dysregulation of androgen secretion. Endocr. Rev. 1995; 16:322-353.
- Gharani N, Waterworth DM, Batty S y col. Association of the steroid synthesis gene CYP11a with polycystic ovary syndrome and hyperandrogenism. Hum. Mol. Genet. 1997; 6:397-402.
- Moran C, Azziz R. The role of suprarrenal cortex in polycystic ovary syndrome. Obstet. Gynecol. Clin. North. Am. 2001; 28:63-75.
- Barnes R, Rosenfield R, Burstein S y col. Pituitary-ovarian responses to nafarelin testing in the polycystic ovary syndrome. N. Engl. J . Med. 1989; 320:559-565.
- Rosenfield RL, Barnes RB, Cara JF y col. Dysregulation of cytochrome P45017 as the cause of polycystic ovarian syndrome. Fertil. Steril. 1990; 53:785-791.
- Fruzzetti F, De Lorenzo D, Ricci C y col. Ovarian influences on suprarrenal androgen secretion in polycystic ovary syndrome. Fertil. Steril. 1995; 63:734-741.
- Gonzalez F, Hatala DA, Speroff L. Suprarrenal and ovarian steroid hormone responses to gonadotropin-releasing hormone agonist treatment in polycystic ovary syndrome. Am. J. Obstet. Gynecol. 1991; 165:535-545.
- Adashi E, Hsueh A, Yen S. Insulin enhacement of luteinizing hormone and follicle-stimulating hormone release by cultured pituitary cells. Endocrinology 1981; 108:1441-1449.
- Yen SSC, Vela P, Rankin J. Inappropiate secretion of follicle stimulating hormone and luteinizing hormone in Polycystic Ovarian Disease. J. Clin. Endocrinol. Metab. 1970; 30:435-442.
- Rebar R, Judd HL, Yen SSC y col. Characterization of the inappropriate gonadotrophin secretion in polycystic ovary syndrome. J. Clin. Endocrinol. Metab. 1976; 57:1320-1329.
- Sir Petermann T. 1997 ¿Modulan los andrógenos la secreción de hormona luteinizante en mujeres hiperandrogénicas. Rev. Med. Chile. 1997; 125:710-718.
- Sir Petermann T, Piwonka V, Perez F y col. Are circulating leptin and luteinizing hormone synchronized in patients with polycystic ovary syndrome. Hum Reprod. 1999; 14:1435-1439.
- Sir Petermann T, Recabarren SE, Lobos A y col. Secretory pattern of leptin and LH during lactational amenorrhoea in breastfeeding normal and polycystic ovarian syndrome women. Hum Reprod. 2001; 16:244-249.
- Sir Petermann T, Devoto L, Maliqueo M y col. Resumption of ovarian function during lactational amenorrhoea in breastfeeding women with polycystic ovary syndrome: endocrine aspects. Hum Reprod. 2001; 16:1603-1610.
- Abbott DH, Dumesic DA, Eisner JR y col. Insights into the development of polycystic ovary syndrome (PCOS) from studies of prenatally androgenized female Rhesus monkeys. Trends Endocrinology and Metabolism. 1998; 9:62-67.
- Zumoff B, Freeman R, Coupey S. A chronobiologic abnormality in luteinizing hormone secretion in teenage girls with the polycystic ovary síndrome. N. Engl. J. Med. 1983; 309:1206-1209.
- Apter D, Butzow T, Laughin GA y col. Accelerated 24 hour luteinizing hormone pulsatile activity in adolescent girls with ovarian hyperandrogenism: relevance to the developmental phase of polycystic ovarian syndrome. J. Clin. Endocrinol. Metab. 1994; 79:119-125.
- Venturoli J, Porcu E, Fabbri R y col. Longitudinal evaluation of the different gonadotropin pulsatile patterns in anovulatory cycles of young girls. J. Clin. Endocrinol. Metab. 1992; 74:836-841.
- Ibáñez L, De Zegher F, Potau N. Anovulation after precocious pubarche: early markers and time course in adolescence. J. Clin. Endocrinol. Metab. 1999; 84:2691-2695.
- Franks S, Robinson S, Willis DS. Nutrition, insulin and polycystic ovary syndrome. J. Reprod. Fertil. 1996; 1:47-53.
- Nestler JE, Jakubowicz DJ. Decreases in ovarian cytochrome P450c17-alpha activity and serum free testosterone after reduction of insulin secretion in polycystic ovary syndrome. N. Engl. J. Med. 1996; 335:617-623.
- Sir Petermann T, Rabenbauer B, Wildt L. The effect of Flutamide on pulsatile gonadotrophin secretion in hyperandrogenemic women. Hum. Reprod. 1993; 8:1807-1812.
- Sir Petermann T, Muñoz A, Candia M y col. LH secretion by the female pituitary: effect of testosterone and the blockade of its receptor. Exp Clin Endocrinol 1996; 104 (Suppl 1):20-22.
- Sir Petermann T, Castillo T, Muñoz S y col. Prueba de tolerancia a la insulina. Un método útil para determinar resistencia insulínica en mujeres hiperandrogénicas obesas. Rev. Méd. Chile. 1996; 124:931-937.
- Sir Petermann T, López G, Castillo T y col. Marcadores bioquímicos y métodos de cuantificación de insulino resistencia en mujeres normales, obesas e hiperandrogénicas. Rev. Med. Chile. 1997; 125:977-985.
- Sir Petermann T, López G, Castillo T y col. Naltrexone effects on insulin sensitivity and insulin secretion in hyperandrogenic women. Exp. Clin. Endocrinol. Diabetes 1998; 106:389-394.
- Burghen GA, Givens JR, Kitabchi AE. Correlation of hyperandrogenism with hyperinsulinemia in polycystic ovarian disease. J. Clin. Endocrinol. Metab. 1980; 50:113-116.
- Nestler JE, Strauss JF III. Insulin as an effector of human ovarian and suprarrenal steroid metabolism. Endocrinol. Metab. Clin. North. Am. 1991; 20:807-832.
- Franks S, Gilling-Smith C, Watson H y col. Insulin action in the normal and polycystic ovary. Endocrinol. Metab. North. Am. 1999; 28:361-378.
- Morimoto S, Fernandez-Mejia C, Romero-Navarro G y col. Testosterone effect on insulin content, messenger ribonucleic acid levels, promoter activity, and secretion in the rat. Endocrinology 2001; 142:1442-1447.
- Dunaif A, Segal KR, Futterweit W y col. Profound peripheral insulin resistance, independent of obesity, in polycystic ovary syndrome. Diabetes 1989; 38:1165-1174.
- Dunaif A, Segal KR, Shelley DR y col. Evidence for distinctive and intrinsic defects in insulin action in polycystic ovary syndrome. Diabetes 1992; 41:1257-1266.
- Dunaif A, Xia J, Book C y col. Excessive insulin receptor serine phosphorilation in cultured fibroblast and skeletal muscle: A potential mechanism for insulin resistance in the polycystic ovary syndrome. J. Clin. Invest. 1995; 96:801-810.
- Dunaif A. Hyperandrogenic anovulation (PCOS): A unique disorder of insulin action associated with an increased risk of non-insulin-dependent diabetes mellitus. Am. J. Med. 1995; 98 (Suppl 1A):33S-39S.
- Barnes RB, Rosenfield RL, Ehrmann DA y col. Ovarian hyperandrogenism as a result of congenital suprarrenal virilizing disorders: evidence for perinatal masculinization of neuroendocrine function in women. J .Clin. Endocrinol. Metab. 1994; 79:1328-1333.
- Clarke IJ, Scaramuzzi RJ, Short RV. Ovulation in prenatal androgenized ewes. J Endocrinol 1977; 73:385-389.
- Dumesic DA, Abbott DH, Eisner JR y col. Prenatal exposure of female rhesus monkeys to testosterone propionate increases serum luteinizing hormone levels in adulthood. Fertil. Steril. 1997; 67:155-163.
- Eisner JR, Dumesic DA, Kemnitz JW y col. Timing of prenatal androgen excess determines differential impairment in insulin secretion and action in adult female rhesus monkeys. J. Clin. Endocrinol. Metab. 2000; 85:1206-1210.
- Eisner JR, Barnett BA, Dumesic DA y col. Ovarian hyperandrogenism in adult female rhesus monkeys exposed to prenatal androgen excess. Fertil Steril 2002; 77:167-172.
- Birch RA, Padmanabhan V, Foster DL y col. Prenatal programming of reproductive neuroendocrine function: fetal androgen exposure produces progressive disruption of reproductive cycles in sheep. Endocrinology 2003; 144:1426-1434.
- Rosser C, Herkimer C, Sarma HN, Recabarren SE y col. Fetal programming: prenatal exposure to excess testosterone programs hyperinsulinemia. Biol Reprod 2003; 68(Suppl 1):293 (Abstract 440).
- Recabarren SE, Sir Petermann T, Lobos A y col. Altered insulin sensitivity indices at early postnatal age following prenatal testosterone exposure. 86th Endocrine Meeting of the Endocrine Society, USA, Abstract P2-73, New Orleans, USA, 16-19 June, 2004
- Sir Petermann T, Maliqueo M, Angel B. Maternal serum androgens in pregnant women with polycystic ovary syndrome: possible implications in prenatal androgenization. Hum Reprod. 2002; 17:2573-2579.
- Kosut SS, Wood RI, Herbosa-Encarnacion C y col. Prenatal androgens time neuroendocrine puberty in the sheep: effect of testosterone dose. Endocrinology 1997; 138:1072-1077.
- Nestler JE. Insulin and insulin-like growht factor I stimulate the 3beta-hydroxisteroid dehydrogenase activity of human placental cytotrophoblast. Endocrinology 1989; 125:2127-2133.
- Nestler JE. Insulin-like growht factor II is a potent inhibitor of the aromatase activity of human placental cytotrophoblast. Endocrinology 1990; 127:2064-2070.
- Abbott DH, Dumesic DA, Franks S. Developmental origin of polycystic ovary syndrome - a hypothesis. J Endocrinol. 2002; 174:1-5.
- Glueck CJ, Goldenberg N, Wang P y col. Metformin during pregnancy reduces insulin, insulin resistance, insulin secretion, weight, testosterone and development of gestational diabetes: prospective longitudinal assessment of women with polycystic ovary syndrome from preconception throughout pregnancy. Hum Reprod. 2004; 19:510-521.
©
Está
expresamente prohibida la redistribución y la redifusión de todo o parte de los
contenidos de la Sociedad Iberoamericana de Información Científica (SIIC) S.A. sin
previo y expreso consentimiento de SIIC



