

-
 Alergia
Alergia -
 Anestesiología
Anestesiología -
 Bioquímica
Bioquímica
-
 Cardiología
Cardiología
-
 Cirugía
Cirugía
-
 Dermatología
Dermatología
-
 Endocrinología y Metabolismo
Endocrinología y Metabolismo
-
 Enfermería
Enfermería
-
 Gastroenterología
Gastroenterología
-
 Hematología
Hematología
-
 Infectología
Infectología
-
 Inmunología
Inmunología
-
 Medicina Interna
Medicina Interna
-
 Nefrología
Nefrología
-
 Neumonología
Neumonología
-
 Neurología
Neurología
-
 Nutrición
Nutrición
-
 Obstetricia y Ginecología
Obstetricia y Ginecología
-
 Odontología
Odontología
-
 Oncología
Oncología
-
 Otorrinolaringología
Otorrinolaringología
-
 Pediatría
Pediatría
-
 Salud Mental
Salud Mental
-
 Salud Pública
Salud Pública
-
 Urología
Urología
-
 Más Especialidades
Más Especialidades
- Índice
-
Acceso por especialidad
-
Alergia
-
Anestesiología
-
Bioquímica
-
Cardiología
-
Cirugía
-
Dermatología
-
Endocrinología y Metabolismo
-
Enfermería
-
Gastroenterología
-
Hematología
-
Infectología
-
Inmunología
-
Medicina Interna
-
Nefrología
-
Neumonología
-
Neurología
-
Nutrición
-
Obstetricia y Ginecología
-
Odontología
-
Oncología
-
Otorrinolaringología
-
Pediatría
-
Salud Mental
-
Salud Pública
-
Urología
-
Más Especialidades
-
Noticias biomédicas
-
Fuentes informativas
- Editoriales
-
COVID-19
-
INFORMES CIENTÍFICOS
- Preguntas con Respuesta
- América Latina Investiga
- Completos revisados (full text)
-
NOTICIAS/OPINIONES
-
Expertos invitados
- Por especialidad
- de Iberoamérica
- del mundo
- Entrevistas
- Casos clínicos
-
Crónicas de Autores
- Authors’ Reports
-
Instrucciones para autores
-
Expertos Preguntan
-
Novedades
- ViASIIC
- Medicina del Dolor
-
Por especialidad
-
Iberomédica
-
Informes comentados
- Textos Completos Autorizados
- Preocupaciones Profesionales
-
Red Científica Iberoamericana (RedCIbe)
-
Conceptos Categoricos
-
Acceso por especialidad

-
Alergia
-
Anestesiología
-
Bioquímica
-
Cardiología
-
Cirugía
-
Dermatología
-
Endocrinología y Metabolismo
-
Enfermería
-
Gastroenterología
-
Hematología
-
Infectología
-
Inmunología
-
Medicina Interna
-
Nefrología
-
Neumonología
-
Neurología
-
Nutrición
-
Obstetricia y Ginecología
-
Odontología
-
Oncología
-
Otorrinolaringología
-
Pediatría
-
Salud Mental
-
Salud Pública
-
Urología
-
Más Especialidades
-
Noticias biomédicas
-
Fuentes informativas
- Editoriales
- Covid-19
-
Informes Científicos
- Preguntas con Respuesta
- América Latina Investiga
- Completos revisados (full text)
- Noticias/Opiniones
- Noticias (castellano/portugués)
- Noticias (otros idiomas)
- Expertos invitados
- Por especialidad
- de Iberoamérica
- del mundo
- Entrevistas
- Casos clínicos
-
Crónicas de Autores
- Authors’ Reports
-
Instrucciones para autores
-
Expertos Preguntan
- Novedades
- ViASIIC
- Medicina del Dolor
-
Por especialidad
-
Iberomédica
-
Informes comentados
- Textos Completos Autorizados
- Preocupaciones Profesionales
- Red Científica Iberoamericana
- Por materia
- Por fecha
-
Conceptos Categóricos

Casos Clínicos
HIPERTENSION ARTERIAL PULMONAR: UNA GRAVE COMPLICACION DE LA ENFERMEDAD DE RENDU-OSLER
Se describe un caso de enfermedad de Rendu Osler que presentó con hipertensión arterial pulmonar, con énfasis en su evolución clínica y manejo terapéutico.
Coautores
Pilar Escribano Subías* Carmen Jiménez López-Guarch* María José Ruiz Cano*
Licenciada en Medicina y Cirugía, Madrid, España*
Pilar Escribano Subías* Carmen Jiménez López-Guarch* María José Ruiz Cano*
Licenciada en Medicina y Cirugía, Madrid, España*
Clasificación en siicsalud
Artículos originales > Expertos del Mundo >
página /dato/casiic.php/87046
Especialidades





Artículos originales > Expertos del Mundo >
página /dato/casiic.php/87046
Especialidades
Primera edición en siicsalud
11 de abril, 2011
11 de abril, 2011
HIPERTENSION ARTERIAL PULMONAR: UNA GRAVE COMPLICACION DE LA ENFERMEDAD DE RENDU-OSLER
(especial para SIIC © Derechos reservados)
(especial para SIIC © Derechos reservados)
La telangiectasia hemorrágica hereditaria (THH) o enfermedad de Rendu-Osler-Weber es una rara displasia vascular con herencia autosómica dominante, penetrancia incompleta y relacionada con la edad. La prevalencia en España se estima en 8.2/100 000 habitantes. Su expresión clínica, muy variable, consiste en la presencia de telangiectasias mucocutáneas y malformaciones arteriovenosas pulmonares, cerebroespinales, hepáticas y en tracto gastrointestinal. El diagnóstico se realiza mediante los criterios de Curaçao1 (Tabla 1).

La epistaxis recurrente es la manifestación más frecuente (90%). Su gravedad aumenta con la edad y las recidivas son frecuentes pese al tratamiento (escleroterapia, láser, cirugía). Hasta el 33% presentan fístulas arteriovenosas pulmonares que en el 50% son sintomáticas: hipoxemia (cortocircuito derecha-izquierda), embolias paradójicas sistémicas (accidentes isquémicos y abscesos cerebrales), hemoptisis y hemotórax. Su mortalidad puede llegar al 22% y se recomienda diagnóstico de cribado y tratamiento, siendo de elección la embolización. Del 9% al 23% presentan malformaciones vasculares cerebroespinales que pueden causar cefalea, epilepsia o hemorragias, aunque la mayoría son asintomáticas. Su detección y tratamiento (microcirugía, radiocirugía estereotáxica, embolización) es recomendable dadas sus devastadoras consecuencias. Las telangiectasias gastrointestinales (estómago y duodeno) aparecen hasta en el 33% de los casos y suelen provocar pérdidas crónicas. El tratamiento es sintomático: hierro y transfusiones. En el caso de hemorragia aguda, la escleroterapia o ablación local pueden ser eficaces a corto plazo, pero no a largo plazo. Son también frecuentes (31%) las malformaciones vasculares hepáticas que, aunque suelen ser asintomáticas, pueden provocar insuficiencia cardíaca de alto gasto, hipertensión portal o afección biliar.1
La TTH es secundaria a mutaciones en 2 genes de la superfamilia de receptores del factor de crecimiento transformante ß (TGF-ß): endoglina (cromosoma 9, TTH tipo 1) y ALK-1 (cromosoma 12, TTH tipo 2). A esta familia también pertenece el gen de la a proteína morfogenética ósea (BMPR-2), ligado a la hipertensión pulmonar (HTP) familiar. La aparición de HTP arterial en estos pacientes se estima en el 15% de los casos y es indistinguible clínica e histológicamente de la HTP idiopática.2,3 Puede deberse a alto gasto cardíaco (secundario a las fístulas arteriovenosas hepáticas) o ser similar a la HTP idiopática. En estos casos, nuevas mutaciones en ALK-1 parecen provocar tanto las dilataciones vasculares características de la TTH como el remodelado de las pequeñas arterias pulmonares que la hacen indistinguible de la HTP idiopática.4
Caso clínico
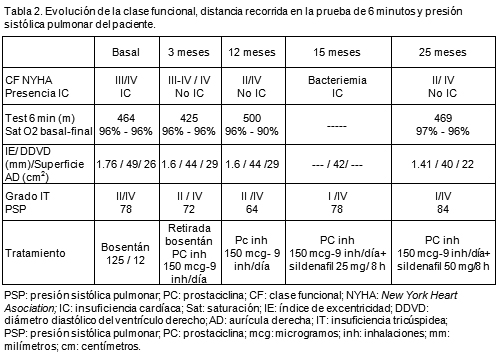
Varón de 30 años, sin antecedentes familiares ni personales de interés, que debutó con hemotórax espontáneo y shock hipovolémico secundario a ruptura de fístula arteriovenosa pulmonar, con requerimientos de lobectomía superior derecha. También presentaba epistaxis recurrentes y telangiectasias en labio inferior y mucosa gastroduodenal, por lo que fue diagnosticado de TTH (criterios de Curaçao1). En los años posteriores presentó frecuentes epistaxis y anemia ferropénica secundaria, precisando transfusiones y escleroterapia nasal en varias ocasiones. A los 43 años se realizó un ecocardiograma por disnea (clasificación funcional II de la New York Heart Association [NYHA]), no atribuible a la anemia, que demostró: ventrículo derecho (VD) dilatado (40 mm), aplanamiento del tabique interventricular, dilatación de tronco pulmonar (39 mm) y presión sistólica pulmonar (PSP) de 90 mm Hg. En su hospital de referencia se realizó estudio etiológico completo de HTP, con resultado negativo y se inició tratamiento convencional.
A la edad de 50 años fue enviado a nuestra Unidad por deterioro progresivo de su clasificación funcional (clase III de la NYHA) e insuficiencia cardíaca, donde, tras descartar otras causas de HTP, se realizó una valoración de la gravedad y estratificación pronóstica. Así, un ecocardiograma puso de manifiesto varios parámetros de mal pronóstico, como VD dilatado e hipocinético, con grave disfunción sistólica, dilatación de aurícula derecha (AD) e índice de excentricidad (IE) elevado. La insuficiencia tricúspidea (IT) era moderada y la presión sistólica pulmonar (PSP) de 78 mm Hg (Tabla 2). La fracción de eyección del VD por ventriculografía isotópica era del 35%. El cateterismo derecho mostró datos de gravedad: AD media: 10 mm Hg; presiones pulmonares: 113/44 mm Hg (media: 64 mm Hg); PCP media: 12 mm Hg; gasto cardíaco: 5.10 l/min, IC 2.7 l/min/m2; resistencias pulmonares arteriolares 10.2 µW. Sin embargo, la distancia recorrida en la prueba de 6 minutos fue aceptable (464 metros) y sin desaturación (saturación basal y final de O2 de 96%). Una tomografía computarizada toracoabdominal demostró gran dilatación de la arteria pulmonar principal (47 mm), con numerosas fístulas arteriovenosas milimétricas pulmonares y hepáticas (figura 1). Mediante ecografía Doppler abdominal se descartó la presencia de hipertensión portal y tampoco se hallaron fístulas arteriovenosas cerebrales en la resonancia magnética. De entre las distintas alternativas terapéuticas optamos por el bosentano y desestimamos la prostaciclina por el riesgo de sangrado inherente al efecto antiagregante.
Dos meses después los niveles de hemoglobina habían descendido 3 puntos, sin datos de sangrado, asociados a presíncopes de esfuerzo. Luego de transfundir sustituimos progresivamente el bosentano por prostaciclina inhalable (150 µg, 9 inhalaciones/día) con mejoría progresiva clínica y analítica. Al año, su CF era II. Había incrementado 40 metros la distancia recorrida en la prueba de 6 minutos y el diámetro diastólico del VD y la presión sistólica pulmonar habían disminuido (Tabla 2) sin fenómenos hemorrágicos. Un mes después ingresó por bacteriemia por Streptococcus viridans, de origen desconocido, con shock séptico, por lo que se practicó infusión de drogas vasoactivas (noradrenalina). Posteriormente, fue necesario asociar sildenafil (50 mg cada 8 h) a la prostaciclina inhalada, para lograr la estabilización del paciente, con buena respuesta clínica durante un seguimiento de 10 meses (Tabla 2).
Aunque la TTH es una de las condiciones relacionadas con la progresión a la HTP, los casos con enfermedad de Rendu-Osler e HTP descritos en la bibliografía no superan la veintena.4-6 La excepcionalidad de nuestro caso se basa en la coincidencia de ambas entidades de forma esporádica (no había antecedentes familiares ni de THH ni de HTP) y en lo tardía que fue la aparición de HTP (13 años tras el diagnóstico de THH) en relación con los casos ya descritos, en los que el diagnóstico de ambas entidades fue simultáneo.5,6 Las manifestaciones de la TTH en nuestro paciente consistían en epistaxis, telangiectasias mucocutáneas y pequeñas fístulas arteriovenosas pulmonares y hepáticas. Estas, por su pequeño tamaño, no pueden explicar la aparición de HTP ni por el alto gasto cardíaco a través del cortocircuito portosistémico ni por la presencia de hipertensión portal.
Si bien no disponemos de estudio genético, la HTP de nuestro paciente podría relacionarse con mutaciones en ALK-1 (gen responsable del tipo 2 de TTH). Hasta la fecha, la correlación genotipo-fenotipo no está del todo establecida, aunque parece que el tipo 1 presenta malformaciones arteriovenosas con mayor frecuencia y gravedad que el tipo 2.1
El tratamiento de estos enfermos tiene ciertas particularidades, ya que la prostaciclina sistémica incrementa el riesgo hemorrágico, ya de por sí elevado.5 El bosentano y el sildenafil se perfilan como alternativas eficaces,7 aunque los vasodilatadores, en conjunto, pueden tener efectos deletéreos al incrementar el shunt arteriovenoso. Dado el importante riesgo hemorrágico de nuestro paciente, iniciamos la terapia con bosentano, cuyo efecto beneficioso ya ha sido descrito aunque con escaso número de sujetos.5,6 Dos meses después sufrió anemia progresiva. Se descartó sangrado digestivo y no hubo epistaxis. El descenso de la hemoglobina es un efecto adverso infrecuente descrito para el bosentano. En general es leve (inferior a 0.9 mg/dl) y suele estabilizarse entre las semanas 4 a 12. Su mecanismo es desconocido (no hay toxicidad medular ni hemólisis) aunque podría estar implicada la hemodilución secundaria a retención de fluidos. Descensos marcados de la hemoglobina, como en nuestro paciente (más de 3 mg/dl) sólo se han descrito en el 0.3% de los casos.8
Nuestra segunda opción fue la prostaciclina inhalable, que cuenta con menos efectos adversos sistémicos,7 con buenos resultados. Nueve meses más tarde se produjo sepsis de origen desconocido con importante deterioro hemodinámico. Fue preciso añadir sildenafil para lograr su estabilización. La asociación de sildenafil como terapia de rescate demostró su eficacia y seguridad a largo plazo en pacientes con deterioro clínico y funcional en tratamiento con prostaciclina en monoterapia.9
El caso que presentamos es doblemente una rareza: en primer lugar, la asociación de TTH con HTP es totalmente esporádica (no existían antecedentes familiares) y la HTP se desencadenó muchos años después del diagnóstico de TTH; en segundo lugar, se produjo un efecto adverso al bosentán, muy infrecuente. Por otro lado, pese al importante riesgo de sangrado de nuestro paciente, no se produjeron fenómenos hemorrágicos con prostaciclina inhalable durante un seguimiento de 18 meses. Por último, hemos de resaltar la importancia del tratamiento combinado, como terapia de rescate, en individuos que presentan deterioro grave con monoterapia.
Los autores no manifiestan "conflictos de interés".
La epistaxis recurrente es la manifestación más frecuente (90%). Su gravedad aumenta con la edad y las recidivas son frecuentes pese al tratamiento (escleroterapia, láser, cirugía). Hasta el 33% presentan fístulas arteriovenosas pulmonares que en el 50% son sintomáticas: hipoxemia (cortocircuito derecha-izquierda), embolias paradójicas sistémicas (accidentes isquémicos y abscesos cerebrales), hemoptisis y hemotórax. Su mortalidad puede llegar al 22% y se recomienda diagnóstico de cribado y tratamiento, siendo de elección la embolización. Del 9% al 23% presentan malformaciones vasculares cerebroespinales que pueden causar cefalea, epilepsia o hemorragias, aunque la mayoría son asintomáticas. Su detección y tratamiento (microcirugía, radiocirugía estereotáxica, embolización) es recomendable dadas sus devastadoras consecuencias. Las telangiectasias gastrointestinales (estómago y duodeno) aparecen hasta en el 33% de los casos y suelen provocar pérdidas crónicas. El tratamiento es sintomático: hierro y transfusiones. En el caso de hemorragia aguda, la escleroterapia o ablación local pueden ser eficaces a corto plazo, pero no a largo plazo. Son también frecuentes (31%) las malformaciones vasculares hepáticas que, aunque suelen ser asintomáticas, pueden provocar insuficiencia cardíaca de alto gasto, hipertensión portal o afección biliar.1
La TTH es secundaria a mutaciones en 2 genes de la superfamilia de receptores del factor de crecimiento transformante ß (TGF-ß): endoglina (cromosoma 9, TTH tipo 1) y ALK-1 (cromosoma 12, TTH tipo 2). A esta familia también pertenece el gen de la a proteína morfogenética ósea (BMPR-2), ligado a la hipertensión pulmonar (HTP) familiar. La aparición de HTP arterial en estos pacientes se estima en el 15% de los casos y es indistinguible clínica e histológicamente de la HTP idiopática.2,3 Puede deberse a alto gasto cardíaco (secundario a las fístulas arteriovenosas hepáticas) o ser similar a la HTP idiopática. En estos casos, nuevas mutaciones en ALK-1 parecen provocar tanto las dilataciones vasculares características de la TTH como el remodelado de las pequeñas arterias pulmonares que la hacen indistinguible de la HTP idiopática.4
Caso clínico
Varón de 30 años, sin antecedentes familiares ni personales de interés, que debutó con hemotórax espontáneo y shock hipovolémico secundario a ruptura de fístula arteriovenosa pulmonar, con requerimientos de lobectomía superior derecha. También presentaba epistaxis recurrentes y telangiectasias en labio inferior y mucosa gastroduodenal, por lo que fue diagnosticado de TTH (criterios de Curaçao1). En los años posteriores presentó frecuentes epistaxis y anemia ferropénica secundaria, precisando transfusiones y escleroterapia nasal en varias ocasiones. A los 43 años se realizó un ecocardiograma por disnea (clasificación funcional II de la New York Heart Association [NYHA]), no atribuible a la anemia, que demostró: ventrículo derecho (VD) dilatado (40 mm), aplanamiento del tabique interventricular, dilatación de tronco pulmonar (39 mm) y presión sistólica pulmonar (PSP) de 90 mm Hg. En su hospital de referencia se realizó estudio etiológico completo de HTP, con resultado negativo y se inició tratamiento convencional.
A la edad de 50 años fue enviado a nuestra Unidad por deterioro progresivo de su clasificación funcional (clase III de la NYHA) e insuficiencia cardíaca, donde, tras descartar otras causas de HTP, se realizó una valoración de la gravedad y estratificación pronóstica. Así, un ecocardiograma puso de manifiesto varios parámetros de mal pronóstico, como VD dilatado e hipocinético, con grave disfunción sistólica, dilatación de aurícula derecha (AD) e índice de excentricidad (IE) elevado. La insuficiencia tricúspidea (IT) era moderada y la presión sistólica pulmonar (PSP) de 78 mm Hg (Tabla 2). La fracción de eyección del VD por ventriculografía isotópica era del 35%. El cateterismo derecho mostró datos de gravedad: AD media: 10 mm Hg; presiones pulmonares: 113/44 mm Hg (media: 64 mm Hg); PCP media: 12 mm Hg; gasto cardíaco: 5.10 l/min, IC 2.7 l/min/m2; resistencias pulmonares arteriolares 10.2 µW. Sin embargo, la distancia recorrida en la prueba de 6 minutos fue aceptable (464 metros) y sin desaturación (saturación basal y final de O2 de 96%). Una tomografía computarizada toracoabdominal demostró gran dilatación de la arteria pulmonar principal (47 mm), con numerosas fístulas arteriovenosas milimétricas pulmonares y hepáticas (figura 1). Mediante ecografía Doppler abdominal se descartó la presencia de hipertensión portal y tampoco se hallaron fístulas arteriovenosas cerebrales en la resonancia magnética. De entre las distintas alternativas terapéuticas optamos por el bosentano y desestimamos la prostaciclina por el riesgo de sangrado inherente al efecto antiagregante.
Dos meses después los niveles de hemoglobina habían descendido 3 puntos, sin datos de sangrado, asociados a presíncopes de esfuerzo. Luego de transfundir sustituimos progresivamente el bosentano por prostaciclina inhalable (150 µg, 9 inhalaciones/día) con mejoría progresiva clínica y analítica. Al año, su CF era II. Había incrementado 40 metros la distancia recorrida en la prueba de 6 minutos y el diámetro diastólico del VD y la presión sistólica pulmonar habían disminuido (Tabla 2) sin fenómenos hemorrágicos. Un mes después ingresó por bacteriemia por Streptococcus viridans, de origen desconocido, con shock séptico, por lo que se practicó infusión de drogas vasoactivas (noradrenalina). Posteriormente, fue necesario asociar sildenafil (50 mg cada 8 h) a la prostaciclina inhalada, para lograr la estabilización del paciente, con buena respuesta clínica durante un seguimiento de 10 meses (Tabla 2).
Aunque la TTH es una de las condiciones relacionadas con la progresión a la HTP, los casos con enfermedad de Rendu-Osler e HTP descritos en la bibliografía no superan la veintena.4-6 La excepcionalidad de nuestro caso se basa en la coincidencia de ambas entidades de forma esporádica (no había antecedentes familiares ni de THH ni de HTP) y en lo tardía que fue la aparición de HTP (13 años tras el diagnóstico de THH) en relación con los casos ya descritos, en los que el diagnóstico de ambas entidades fue simultáneo.5,6 Las manifestaciones de la TTH en nuestro paciente consistían en epistaxis, telangiectasias mucocutáneas y pequeñas fístulas arteriovenosas pulmonares y hepáticas. Estas, por su pequeño tamaño, no pueden explicar la aparición de HTP ni por el alto gasto cardíaco a través del cortocircuito portosistémico ni por la presencia de hipertensión portal.
Si bien no disponemos de estudio genético, la HTP de nuestro paciente podría relacionarse con mutaciones en ALK-1 (gen responsable del tipo 2 de TTH). Hasta la fecha, la correlación genotipo-fenotipo no está del todo establecida, aunque parece que el tipo 1 presenta malformaciones arteriovenosas con mayor frecuencia y gravedad que el tipo 2.1
El tratamiento de estos enfermos tiene ciertas particularidades, ya que la prostaciclina sistémica incrementa el riesgo hemorrágico, ya de por sí elevado.5 El bosentano y el sildenafil se perfilan como alternativas eficaces,7 aunque los vasodilatadores, en conjunto, pueden tener efectos deletéreos al incrementar el shunt arteriovenoso. Dado el importante riesgo hemorrágico de nuestro paciente, iniciamos la terapia con bosentano, cuyo efecto beneficioso ya ha sido descrito aunque con escaso número de sujetos.5,6 Dos meses después sufrió anemia progresiva. Se descartó sangrado digestivo y no hubo epistaxis. El descenso de la hemoglobina es un efecto adverso infrecuente descrito para el bosentano. En general es leve (inferior a 0.9 mg/dl) y suele estabilizarse entre las semanas 4 a 12. Su mecanismo es desconocido (no hay toxicidad medular ni hemólisis) aunque podría estar implicada la hemodilución secundaria a retención de fluidos. Descensos marcados de la hemoglobina, como en nuestro paciente (más de 3 mg/dl) sólo se han descrito en el 0.3% de los casos.8
Nuestra segunda opción fue la prostaciclina inhalable, que cuenta con menos efectos adversos sistémicos,7 con buenos resultados. Nueve meses más tarde se produjo sepsis de origen desconocido con importante deterioro hemodinámico. Fue preciso añadir sildenafil para lograr su estabilización. La asociación de sildenafil como terapia de rescate demostró su eficacia y seguridad a largo plazo en pacientes con deterioro clínico y funcional en tratamiento con prostaciclina en monoterapia.9
El caso que presentamos es doblemente una rareza: en primer lugar, la asociación de TTH con HTP es totalmente esporádica (no existían antecedentes familiares) y la HTP se desencadenó muchos años después del diagnóstico de TTH; en segundo lugar, se produjo un efecto adverso al bosentán, muy infrecuente. Por otro lado, pese al importante riesgo de sangrado de nuestro paciente, no se produjeron fenómenos hemorrágicos con prostaciclina inhalable durante un seguimiento de 18 meses. Por último, hemos de resaltar la importancia del tratamiento combinado, como terapia de rescate, en individuos que presentan deterioro grave con monoterapia.
Los autores no manifiestan "conflictos de interés".
Ángela Flox Camacho, Hospital Universitario "12 de Octubre" Unidad de Hipertensión Pulmonar, Insufiencia Cardíaca y Trasplante Cardíaco, 28032, Madrid, España,
e-mail: angelaflox@gmail.com
1. Pérez de Molino A, Zarrabeitia R, Fernández A. Telangiectasia hemorrágica hereditaria. Med Clin 124(15):583-7, 2005.
2. Simonneau G, Galiè N, Rubin LJ, Langleben D, Seeger W, Domenighetti G, et al. Clinical classification of pulmonary hypertension. J Am Coll Cardiol 43(6):5S-12S, 2005.
3. Farber HW, Loscalzo J. Mechanisms of disease: pulmonary arterial hypertension. N Engl J Med 351:1655-65, 2004.
4. Trembath R, Thomson J, Machado R, Morgan N, Atkinson C, Winship I, et al. Clinical and molecular genetic features of pulmonary hypertension in patients with hereditary haemorrhagic telangiectasia. N Engl J Med 345:325-34, 2001.
5. Schalg K, Opitz C,Wensel R, Felix S, Ewert R. Pulmonary hipertensión in hereditary haemorrhagic teleangiectasia (Rendu-Osler-Weber disease). Progression over 10 years. Dtsch Med Wochenschr 130(23):1434-7, 2005.
6. Nowotny R, Bonderman D, Skoro-Sajer N, Jakowitsch J, Adlbrecht C, Lang IM. Bosentan therapy in pulmonary arterial hypertension associated with hereditary haemorrhagic telangiectasia [abstract]. ESC Congress Stockholm, 2005.
7. Galiè N, Torbicki A, Barst R, Datervelle P, Haworth S, Higenbottan T, et al. Guías de la práctica clínica sobre el diagnóstico y tratamiento de la hipertensión arterial pulmonar. Rev Esp Cardiol 58(5):523-66, 2005.
8. Escribano Subías P. Tratamiento convencional y farmacológico oral. En: Ministerio de Sanidad y Consumo, Organización Médica Colegial, editores. Evidencia científica en Hipertensión arterial pulmonar. Manual de actuación. Madrid: IM&C, SA, p. 58, 2006.
9. Jiménez López-Guarch C, Escribano Subías P, Tello de Meneses R, Delgado Jiménez JF, Sadia Pérez D, Velásquez Martín MT, et al. Eficacia del sildenafilo por vía oral como terapia de rescate en pacientes con hipertensión arterial pulmonar severa en tratamiento crónico con prostaciclina. Resultados a largo plazo. Rev Esp Cardiol 57(10):946-51, 2004.
Está expresamente prohibida la redistribución y la redifusión de todo o parte de los
contenidos de la Sociedad Iberoamericana de Información Científica (SIIC) S.A. sin
previo y expreso consentimiento de SIIC.
ua91218

