

-
 Alergia
Alergia -
 Anestesiología
Anestesiología -
 Bioquímica
Bioquímica
-
 Cardiología
Cardiología
-
 Cirugía
Cirugía
-
 Dermatología
Dermatología
-
 Endocrinología y Metabolismo
Endocrinología y Metabolismo
-
 Enfermería
Enfermería
-
 Gastroenterología
Gastroenterología
-
 Hematología
Hematología
-
 Infectología
Infectología
-
 Inmunología
Inmunología
-
 Medicina Interna
Medicina Interna
-
 Nefrología
Nefrología
-
 Neumonología
Neumonología
-
 Neurología
Neurología
-
 Nutrición
Nutrición
-
 Obstetricia y Ginecología
Obstetricia y Ginecología
-
 Odontología
Odontología
-
 Oncología
Oncología
-
 Otorrinolaringología
Otorrinolaringología
-
 Pediatría
Pediatría
-
 Salud Mental
Salud Mental
-
 Salud Pública
Salud Pública
-
 Urología
Urología
-
 Más Especialidades
Más Especialidades
- Índice
-
Acceso por especialidad
-
Alergia
-
Anestesiología
-
Bioquímica
-
Cardiología
-
Cirugía
-
Dermatología
-
Endocrinología y Metabolismo
-
Enfermería
-
Gastroenterología
-
Hematología
-
Infectología
-
Inmunología
-
Medicina Interna
-
Nefrología
-
Neumonología
-
Neurología
-
Nutrición
-
Obstetricia y Ginecología
-
Odontología
-
Oncología
-
Otorrinolaringología
-
Pediatría
-
Salud Mental
-
Salud Pública
-
Urología
-
Más Especialidades
-
Noticias biomédicas
-
Fuentes informativas
- Editoriales
-
COVID-19
-
INFORMES CIENTÍFICOS
- Preguntas con Respuesta
- América Latina Investiga
- Completos revisados (full text)
-
NOTICIAS/OPINIONES
-
Expertos invitados
- Por especialidad
- de Iberoamérica
- del mundo
- Entrevistas
- Casos clínicos
-
Crónicas de Autores
- Authors’ Reports
-
Instrucciones para autores
-
Expertos Preguntan
-
Novedades
- ViASIIC
- Medicina del Dolor
-
Por especialidad
-
Iberomédica
-
Informes comentados
- Textos Completos Autorizados
- Preocupaciones Profesionales
-
Red Científica Iberoamericana (RedCIbe)
-
Conceptos Categoricos
-
Acceso por especialidad

-
Alergia
-
Anestesiología
-
Bioquímica
-
Cardiología
-
Cirugía
-
Dermatología
-
Endocrinología y Metabolismo
-
Enfermería
-
Gastroenterología
-
Hematología
-
Infectología
-
Inmunología
-
Medicina Interna
-
Nefrología
-
Neumonología
-
Neurología
-
Nutrición
-
Obstetricia y Ginecología
-
Odontología
-
Oncología
-
Otorrinolaringología
-
Pediatría
-
Salud Mental
-
Salud Pública
-
Urología
-
Más Especialidades
-
Noticias biomédicas
-
Fuentes informativas
- Editoriales
- Covid-19
-
Informes Científicos
- Preguntas con Respuesta
- América Latina Investiga
- Completos revisados (full text)
- Noticias/Opiniones
- Noticias (castellano/portugués)
- Noticias (otros idiomas)
- Expertos invitados
- Por especialidad
- de Iberoamérica
- del mundo
- Entrevistas
- Casos clínicos
-
Crónicas de Autores
- Authors’ Reports
-
Instrucciones para autores
-
Expertos Preguntan
- Novedades
- ViASIIC
- Medicina del Dolor
-
Por especialidad
-
Iberomédica
-
Informes comentados
- Textos Completos Autorizados
- Preocupaciones Profesionales
- Red Científica Iberoamericana
- Por materia
- Por fecha
-
Conceptos Categóricos

Casos Clínicos
UN DILEMA DIAGNOSTICO EN UN PACIENTE CON LINFOMA DE LENTA EVOLUCION Y LA APARICION DE UN SARCOMA HISTIOCITICO DE GRAN MALIGNIDAD
La sospecha de un sarcoma histiocítico obliga al diagnóstico diferencial con neoplasias de origen linfático, por lo cual resulta esencial la aplicación de técnicas de inmunohistoquímica.
Coautores
Mariano Adrián Forlino*
Médico Especialista en Medicina Interna, Jefe de Trabajos Prácticos de Farmacología de la Facultad de Medicina de la Universidad de Buenos Aires, Buenos Aires, Argentina*
Mariano Adrián Forlino*
Médico Especialista en Medicina Interna, Jefe de Trabajos Prácticos de Farmacología de la Facultad de Medicina de la Universidad de Buenos Aires, Buenos Aires, Argentina*
Clasificación en siicsalud
Artículos originales > Expertos del Mundo >
página /dato/casiic.php/86828
Especialidades



Artículos originales > Expertos del Mundo >
página /dato/casiic.php/86828
Especialidades
Primera edición en siicsalud
30 de marzo, 2010
30 de marzo, 2010
UN DILEMA DIAGNOSTICO EN UN PACIENTE CON LINFOMA DE LENTA EVOLUCION Y LA APARICION DE UN SARCOMA HISTIOCITICO DE GRAN MALIGNIDAD
(especial para SIIC © Derechos reservados)
(especial para SIIC © Derechos reservados)
Introducción
El sarcoma histiocítico es una neoplasia caracterizada por la proliferación maligna de células con morfología e inmunofenotipo similares a los de los histiocitos maduros. Se trata de una enfermedad infrecuente, que afecta en especial a pacientes adultos de sexo masculino. Su etiología es desconocida y compromete los ganglios linfáticos en el 30% de los enfermos, en otro 30% a la piel y en el 30% de los casos abarca sitios extranodales.
Caso clínico
Paciente varón, de 82 años, que consultó 15 años atrás por una lesión de 4 mm en la piel del tronco que, por rascado, le había producido un leve sangrado. El examen físico en ese momento reveló una hepatomegalia firme, de 4 cm bajo el reborde costal y un polo de bazo palpable. El resto del examen físico no brindó anormalidades. Los exámenes de laboratorio fueron normales, excepto por la presencia de una banda monoclonal de IgG, con eritrosedimentación (ESD) de 80 mm/h que presentaba desde 3 años atrás, por lo que había sido estudiado por otro colega, descartando un mieloma múltiple. Esa banda monoclonal se había incrementado de 2 g en un principio hasta 3.5 g en la nueva consulta, al igual que la ESD (50 mm/h en la primera evaluación y 80 mm/h en la segunda). El hematocrito era de 42%.
En la tomografía computarizada (TAC) de cuerpo entero sólo se observó la hepatomegalia homogénea y el ligero agrandamiento esplénico. El paciente estaba totalmente asintomático, dirigía diariamente su actividad empresaria y 2 días por semana jugaba al golf. Mediante una biopsia de piel se diagnosticó un epitelioma basocelular que fue extirpado.
Se había decidido 15 años antes la realización de una biopsia hepática percutánea, con la cual se hizo el diagnóstico de linfoma B de bajo grado. De acuerdo con el servicio de Hematología se resolvió no efectuar tratamiento oncológico, dada las características de esta neoplasia en un paciente asintomático y que realizaba una vida normal.
Concurría a la consulta cada 3 meses. La ESD continuó en ascenso, hasta llegar a 120 mm/h. Siete años después del diagnóstico, el enfermo padeció un accidente cerebrovascular (ACV) isquémico leve, con recuperación total. Fue medicado con ticlopidina en forma permanente.
Un año después, en un estudio de laboratorio se comprobó una elevación del antígeno prostático específico. Mediante una ecografía transrectal con biopsias se efectuó el diagnóstico de un adenocarcinoma de próstata intracapsular, por lo que se realizó la resección quirúrgica. Sin embargo, surgieron dificultades con el servicio de Urología para decidir la cirugía de un paciente con una ESD de 120 mm/h. El enfermo siguió su vida normal, con controles periódicos clínicos y de laboratorio. Presentaba molestias ligeras, que se atribuían a la edad y no a un compromiso sistémico. En este contexto, relataba la presencia de gonalgias relacionadas con la práctica de 18 hoyos de golf, por lo que se le recomendó la reducción de la actividad a la mitad. Un año después, en un viaje a China, presentó un grave episodio de gastroenteritis (diarrea del viajero), tratada de manera adecuada por el médico del grupo. Durante todos esos años le fueron extirpados 5 epiteliomas basocelulares. En la última consulta de control, se detectó una tumoración dolorosa, palpable y visible en el centro del cráneo. El aspecto era francamente neoplásico, con bordes irregulares y zonas necróticas alternadas con franjas leñosas (figura 1).
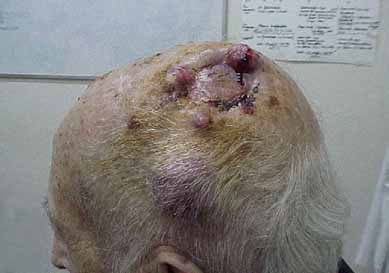
Se procedió a la exéresis para estudio histopatológico. En espera del resultado se efectuaron una fibroendoscopia digestiva alta, una fibrocolonoscopia y una TAC de cuerpo entero que incluyó el sistema nervioso central. Se descartó compromiso del aparato digestivo, dado que había surgido anemia en poco tiempo.
En las imágenes no se demostró compromiso óseo de la formación isodensa, aunque se observaron ganglios peripancreáticos, retroperitoneales e inguinales, que ya presentaba en estudios anteriores y eran menores de 2 cm. Para disminuir el dolor se inició radioterapia, sin ningún beneficio. Se efectuaba tratamiento y curas locales en el Servicio de Cirugía Plástica. El tumor siguió avanzando notoriamente día a día, llegando a cubrir toda la calota y región frontal, acercándose a la órbita izquierda. Surgió una formación de gran tamaño en la zona izquierda del cuello, muy dolorosa, que alcanzó el tamaño de una naranja y obligaba al paciente a mantener una posición inclinada de la cabeza.
En la anatomía patológica (figura 2) se informó proliferación de grandes células, de núcleos extensos vesiculares, con nucleolos destacados acidófilos, de forma ovoide o polilobulada con multinucleación en la periferia celular, excéntricos, citoplasma abundante acidófilo, homogéneo o microvacuolar. El índice mitótico era muy elevado, con mitosis típicas o atípicas, además de focos de necrosis. Presentaba pobre cohesión celular, con infiltración de dermis y tejidos mesenquimáticos subyacentes y erosión de la epidermis. En la inmunomarcación se mostró negatividad para anticuerpos pancitoqueratina y antígeno carcinoembrionario, con lo que se descartó epitelioma o adenocarcinoma. La negatividad para anticuerpos CD30, CD3 y TIA1 permitió descartar linfoma anaplásico de células tipo T, linfoma inmunoblástico T y de células NK. También fueron negativos los anticuerpos CD34 y actina músculo-específica, con lo que se desestimó el diagnóstico de sarcoma mieloide, angiosarcoma y sarcoma epitelioide. Finalmente fueron positivos los anticuerpos para vimentina (demostración de sarcoma) y CD68 (estirpe histiocítica) (figura 2)..

Discusión
La bibliografía disponible acerca de esta enfermedad se refiere habitualmente a revisiones de casos realizadas por medio de técnicas inmunohistoquímicas. En la mayoría de las casuísticas se describe una enfermedad rápidamente evolutiva y fatal, con exámenes histopatológicos marcadamente pleomórficos en cuanto a su celularidad, y que sólo pueden diferenciarse de otras entidades tales como el linfoma T de células grandes, el sarcoma de células de Langerhans o el sarcoma de células foliculares dendríticas, teniendo en cuenta como fundamento a la inmunohistoquímica. En el caso de este paciente, observamos una enfermedad maligna de rápida evolución, con poca sintomatología sistémica, cuyo diagnóstico sólo pudo efectuarse mediante de un minucioso examen histopatológico con un extenso panel de inmunohistoquímica.
Este caso resulta de interés por 2 motivos: la infrecuente situación de un paciente portador de una enfermedad neoplásica (linfoma) que pudo ser controlado durante 15 años por el mismo médico, con visitas periódicas que cumplía adecuadamente, con las intercurrencias propias de un enfermo añoso (cáncer de próstata, ACV), que trabajó hasta 2 meses antes de su deceso; y el otro motivo, que un parsimonioso linfoma de 15 años de evolución, se transformara en un infrecuente sarcoma histiocítico de piel. Al considerar que el sarcoma histiocítico es una neoplasia caracterizada por la proliferación maligna de células con morfología e inmunofenotipo similares a los histiocitos maduros, la sospecha ante un caso de estas características obliga a discernir entre distintas neoplasias de origen linfático como diagnóstico diferencial, por lo que es imprescindible un adecuado diagnóstico inmunohistoquímico.
El sarcoma histiocítico es una neoplasia caracterizada por la proliferación maligna de células con morfología e inmunofenotipo similares a los de los histiocitos maduros. Se trata de una enfermedad infrecuente, que afecta en especial a pacientes adultos de sexo masculino. Su etiología es desconocida y compromete los ganglios linfáticos en el 30% de los enfermos, en otro 30% a la piel y en el 30% de los casos abarca sitios extranodales.
Caso clínico
Paciente varón, de 82 años, que consultó 15 años atrás por una lesión de 4 mm en la piel del tronco que, por rascado, le había producido un leve sangrado. El examen físico en ese momento reveló una hepatomegalia firme, de 4 cm bajo el reborde costal y un polo de bazo palpable. El resto del examen físico no brindó anormalidades. Los exámenes de laboratorio fueron normales, excepto por la presencia de una banda monoclonal de IgG, con eritrosedimentación (ESD) de 80 mm/h que presentaba desde 3 años atrás, por lo que había sido estudiado por otro colega, descartando un mieloma múltiple. Esa banda monoclonal se había incrementado de 2 g en un principio hasta 3.5 g en la nueva consulta, al igual que la ESD (50 mm/h en la primera evaluación y 80 mm/h en la segunda). El hematocrito era de 42%.
En la tomografía computarizada (TAC) de cuerpo entero sólo se observó la hepatomegalia homogénea y el ligero agrandamiento esplénico. El paciente estaba totalmente asintomático, dirigía diariamente su actividad empresaria y 2 días por semana jugaba al golf. Mediante una biopsia de piel se diagnosticó un epitelioma basocelular que fue extirpado.
Se había decidido 15 años antes la realización de una biopsia hepática percutánea, con la cual se hizo el diagnóstico de linfoma B de bajo grado. De acuerdo con el servicio de Hematología se resolvió no efectuar tratamiento oncológico, dada las características de esta neoplasia en un paciente asintomático y que realizaba una vida normal.
Concurría a la consulta cada 3 meses. La ESD continuó en ascenso, hasta llegar a 120 mm/h. Siete años después del diagnóstico, el enfermo padeció un accidente cerebrovascular (ACV) isquémico leve, con recuperación total. Fue medicado con ticlopidina en forma permanente.
Un año después, en un estudio de laboratorio se comprobó una elevación del antígeno prostático específico. Mediante una ecografía transrectal con biopsias se efectuó el diagnóstico de un adenocarcinoma de próstata intracapsular, por lo que se realizó la resección quirúrgica. Sin embargo, surgieron dificultades con el servicio de Urología para decidir la cirugía de un paciente con una ESD de 120 mm/h. El enfermo siguió su vida normal, con controles periódicos clínicos y de laboratorio. Presentaba molestias ligeras, que se atribuían a la edad y no a un compromiso sistémico. En este contexto, relataba la presencia de gonalgias relacionadas con la práctica de 18 hoyos de golf, por lo que se le recomendó la reducción de la actividad a la mitad. Un año después, en un viaje a China, presentó un grave episodio de gastroenteritis (diarrea del viajero), tratada de manera adecuada por el médico del grupo. Durante todos esos años le fueron extirpados 5 epiteliomas basocelulares. En la última consulta de control, se detectó una tumoración dolorosa, palpable y visible en el centro del cráneo. El aspecto era francamente neoplásico, con bordes irregulares y zonas necróticas alternadas con franjas leñosas (figura 1).
Se procedió a la exéresis para estudio histopatológico. En espera del resultado se efectuaron una fibroendoscopia digestiva alta, una fibrocolonoscopia y una TAC de cuerpo entero que incluyó el sistema nervioso central. Se descartó compromiso del aparato digestivo, dado que había surgido anemia en poco tiempo.
En las imágenes no se demostró compromiso óseo de la formación isodensa, aunque se observaron ganglios peripancreáticos, retroperitoneales e inguinales, que ya presentaba en estudios anteriores y eran menores de 2 cm. Para disminuir el dolor se inició radioterapia, sin ningún beneficio. Se efectuaba tratamiento y curas locales en el Servicio de Cirugía Plástica. El tumor siguió avanzando notoriamente día a día, llegando a cubrir toda la calota y región frontal, acercándose a la órbita izquierda. Surgió una formación de gran tamaño en la zona izquierda del cuello, muy dolorosa, que alcanzó el tamaño de una naranja y obligaba al paciente a mantener una posición inclinada de la cabeza.
En la anatomía patológica (figura 2) se informó proliferación de grandes células, de núcleos extensos vesiculares, con nucleolos destacados acidófilos, de forma ovoide o polilobulada con multinucleación en la periferia celular, excéntricos, citoplasma abundante acidófilo, homogéneo o microvacuolar. El índice mitótico era muy elevado, con mitosis típicas o atípicas, además de focos de necrosis. Presentaba pobre cohesión celular, con infiltración de dermis y tejidos mesenquimáticos subyacentes y erosión de la epidermis. En la inmunomarcación se mostró negatividad para anticuerpos pancitoqueratina y antígeno carcinoembrionario, con lo que se descartó epitelioma o adenocarcinoma. La negatividad para anticuerpos CD30, CD3 y TIA1 permitió descartar linfoma anaplásico de células tipo T, linfoma inmunoblástico T y de células NK. También fueron negativos los anticuerpos CD34 y actina músculo-específica, con lo que se desestimó el diagnóstico de sarcoma mieloide, angiosarcoma y sarcoma epitelioide. Finalmente fueron positivos los anticuerpos para vimentina (demostración de sarcoma) y CD68 (estirpe histiocítica) (figura 2)..
Discusión
La bibliografía disponible acerca de esta enfermedad se refiere habitualmente a revisiones de casos realizadas por medio de técnicas inmunohistoquímicas. En la mayoría de las casuísticas se describe una enfermedad rápidamente evolutiva y fatal, con exámenes histopatológicos marcadamente pleomórficos en cuanto a su celularidad, y que sólo pueden diferenciarse de otras entidades tales como el linfoma T de células grandes, el sarcoma de células de Langerhans o el sarcoma de células foliculares dendríticas, teniendo en cuenta como fundamento a la inmunohistoquímica. En el caso de este paciente, observamos una enfermedad maligna de rápida evolución, con poca sintomatología sistémica, cuyo diagnóstico sólo pudo efectuarse mediante de un minucioso examen histopatológico con un extenso panel de inmunohistoquímica.
Este caso resulta de interés por 2 motivos: la infrecuente situación de un paciente portador de una enfermedad neoplásica (linfoma) que pudo ser controlado durante 15 años por el mismo médico, con visitas periódicas que cumplía adecuadamente, con las intercurrencias propias de un enfermo añoso (cáncer de próstata, ACV), que trabajó hasta 2 meses antes de su deceso; y el otro motivo, que un parsimonioso linfoma de 15 años de evolución, se transformara en un infrecuente sarcoma histiocítico de piel. Al considerar que el sarcoma histiocítico es una neoplasia caracterizada por la proliferación maligna de células con morfología e inmunofenotipo similares a los histiocitos maduros, la sospecha ante un caso de estas características obliga a discernir entre distintas neoplasias de origen linfático como diagnóstico diferencial, por lo que es imprescindible un adecuado diagnóstico inmunohistoquímico.
Gerardo Manuel Baré, 1425, Buenos Aires, Argentina,
e-mail: gbare@ciudad.com.ar
Lauritzen AF, Delsol G, Hansen NE, Horn T, Ersboll J, Hou-Jensen K, Ralfkiaer E. Am J Clin Pathol. 1994 Jul;102(1):45-54.
Pileri SA, Grogan TM, Harris NL, Banks P, Campo E, Chan JK, Favera RD, et al. Histopathology. 2002 Jul;41(1):1-29.
Soria C, Orradre JL, Garcia-Almagro D, Martinez B, Algara P, Piris MA. Am J Dermatopathol. 1992 Dec;14(6):511-7.
Hanson CA, Jaszcz W, Kersey JH, Astorga MG, Peterson BA, Gajl-Peczalska KJ, Frizzera G. Br J Haematol. 1989 Oct;73(2):187-98.
Lauritzen AF, Ralfkiaer E. Leuk Lymphoma. 1995 Jun;18(1-2):73-80.
Copie-Bergman C, Wotherspoon AC, Norton AJ, Diss TC, Isaacson PG. Am J Surg Pathol. 1998 Nov;22(11):1386-92.
Está expresamente prohibida la redistribución y la redifusión de todo o parte de los
contenidos de la Sociedad Iberoamericana de Información Científica (SIIC) S.A. sin
previo y expreso consentimiento de SIIC.
ua91218

