

-
 Alergia
Alergia -
 Anestesiología
Anestesiología -
 Bioquímica
Bioquímica
-
 Cardiología
Cardiología
-
 Cirugía
Cirugía
-
 Dermatología
Dermatología
-
 Endocrinología y Metabolismo
Endocrinología y Metabolismo
-
 Enfermería
Enfermería
-
 Gastroenterología
Gastroenterología
-
 Hematología
Hematología
-
 Infectología
Infectología
-
 Inmunología
Inmunología
-
 Medicina Interna
Medicina Interna
-
 Nefrología
Nefrología
-
 Neumonología
Neumonología
-
 Neurología
Neurología
-
 Nutrición
Nutrición
-
 Obstetricia y Ginecología
Obstetricia y Ginecología
-
 Odontología
Odontología
-
 Oncología
Oncología
-
 Otorrinolaringología
Otorrinolaringología
-
 Pediatría
Pediatría
-
 Salud Mental
Salud Mental
-
 Salud Pública
Salud Pública
-
 Urología
Urología
-
 Más Especialidades
Más Especialidades
- Índice
-
Acceso por especialidad
-
Alergia
-
Anestesiología
-
Bioquímica
-
Cardiología
-
Cirugía
-
Dermatología
-
Endocrinología y Metabolismo
-
Enfermería
-
Gastroenterología
-
Hematología
-
Infectología
-
Inmunología
-
Medicina Interna
-
Nefrología
-
Neumonología
-
Neurología
-
Nutrición
-
Obstetricia y Ginecología
-
Odontología
-
Oncología
-
Otorrinolaringología
-
Pediatría
-
Salud Mental
-
Salud Pública
-
Urología
-
Más Especialidades
-
Noticias biomédicas
-
Fuentes informativas
- Editoriales
-
COVID-19
-
INFORMES CIENTÍFICOS
- Preguntas con Respuesta
- América Latina Investiga
- Completos revisados (full text)
-
NOTICIAS/OPINIONES
-
Expertos invitados
- Por especialidad
- de Iberoamérica
- del mundo
- Entrevistas
- Casos clínicos
-
Crónicas de Autores
- Authors’ Reports
-
Instrucciones para autores
-
Expertos Preguntan
-
Novedades
- ViASIIC
- Medicina del Dolor
-
Por especialidad
-
Iberomédica
-
Informes comentados
- Textos Completos Autorizados
- Preocupaciones Profesionales
-
Red Científica Iberoamericana (RedCIbe)
-
Conceptos Categoricos
-
Acceso por especialidad

-
Alergia
-
Anestesiología
-
Bioquímica
-
Cardiología
-
Cirugía
-
Dermatología
-
Endocrinología y Metabolismo
-
Enfermería
-
Gastroenterología
-
Hematología
-
Infectología
-
Inmunología
-
Medicina Interna
-
Nefrología
-
Neumonología
-
Neurología
-
Nutrición
-
Obstetricia y Ginecología
-
Odontología
-
Oncología
-
Otorrinolaringología
-
Pediatría
-
Salud Mental
-
Salud Pública
-
Urología
-
Más Especialidades
-
Noticias biomédicas
-
Fuentes informativas
- Editoriales
- Covid-19
-
Informes Científicos
- Preguntas con Respuesta
- América Latina Investiga
- Completos revisados (full text)
- Noticias/Opiniones
- Noticias (castellano/portugués)
- Noticias (otros idiomas)
- Expertos invitados
- Por especialidad
- de Iberoamérica
- del mundo
- Entrevistas
- Casos clínicos
-
Crónicas de Autores
- Authors’ Reports
-
Instrucciones para autores
-
Expertos Preguntan
- Novedades
- ViASIIC
- Medicina del Dolor
-
Por especialidad
-
Iberomédica
-
Informes comentados
- Textos Completos Autorizados
- Preocupaciones Profesionales
- Red Científica Iberoamericana
- Por materia
- Por fecha
-
Conceptos Categóricos

Casos Clínicos
MIELOMA MULTIPLE NO SECRETOR
El mieloma no secretor representa menos del 1% de todos los mielomas. Su peculiaridad reside en la ausencia de componente monoclonal tanto en suero como en orina. Se presenta un caso de la enfermedad de difícil diagnóstico.
Coautores
Patricia Gracia García* Rocio Zamora González-Mariño** Juan Ignacio Pérez Calvo***
Médico interno residente de psiquiatría, Zaragoza, España*
Médico interno residente de nefrología, Zaragoza, España**
Jefe de sección medicina interna, Profesor asociado médico, Zaragoza, España***
Patricia Gracia García* Rocio Zamora González-Mariño** Juan Ignacio Pérez Calvo***
Médico interno residente de psiquiatría, Zaragoza, España*
Médico interno residente de nefrología, Zaragoza, España**
Jefe de sección medicina interna, Profesor asociado médico, Zaragoza, España***
Clasificación en siicsalud
Artículos originales > Expertos del Mundo >
página /dato/casiic.php/86775
Especialidades







Artículos originales > Expertos del Mundo >
página /dato/casiic.php/86775
Especialidades
Primera edición en siicsalud
18 de agosto, 2010
18 de agosto, 2010
Introducción
El mieloma no secretor representa menos del 1% de todos los mielomas. Clínicamente, hematológicamente y bioquímicamente los hallazgos son similares a los encontrados en el mieloma secretor. Su peculiaridad reside en la ausencia de componente monoclonal tanto en suero como en orina. El diagnóstico puede retrasarse dado que en forma inicial presenta un patrón clínico muy inespecífico, especialmente aquellos casos en que los estudios radiológicos no revelan lesiones líticas. Se presenta un caso de mieloma múltiple no secretor de difícil diagnóstico.
Caso clínico
Se trata de un varón de 72 años con antecedentes personales de diabetes mellitus tipo 2, traumatismo tras caída accidental con aplastamiento vertebral a nivel de L1 y fractura de cóccix secundaria, discopatía degenerativa y distimia.
Ingresa en nuestro servicio procedente de consultas externas, para diagnóstico y tratamiento de lumbociatalgia bilateral progresiva de predominio izquierdo asociada con cervicobraquialgia derecha, de aproximadamente 8 meses de evolución y de características inflamatorias. No presenta clínica sistémica. En la exploración física presenta un buen estado general, destaca un soplo sistólico en foco aórtico a la auscultación cardíaca y, neurológicamente, abolición de reflejos osteotendinosos aquíleos y patelares bilaterales e hipoestesia táctil en cara externa de pierna izquierda. No se observa déficit motor. En el tacto rectal: próstata de aspecto adenomatoso de grado II.
A su ingreso se le realiza una serie de pruebas complementarias básicas que incluyen perfil analítico general y radiografías. En el hemograma destaca una anemia normocítica leve (hemoglobina = 11 mg/dl) con una velocidad de sedimentación globular (VSG) de 13 mm. En la bioquímica (incluido el perfil hepático y lipídico) no existen hallazgos patológicos, presenta niveles de urea y creatinina de 19 mg/dl y 0.6 mg/dl, respectivamente, y calcemia de 9.52 mg/dl con fosfatemia de 4.46 mg/dl. El proteinograma muestra un descenso de proteínas (5.5 g/dl) a expensas de las gammaglobulinas (7.1%).
En la radiografía de tórax se observan signos de enfisema pulmonar y osteólisis en húmero derecho y ambas clavículas. En la radiografía simple de columna vertebral lumbar presenta osteoporosis difusa con acuñamientos vertebrales D9-D12 y patrón permeable L3-L5.
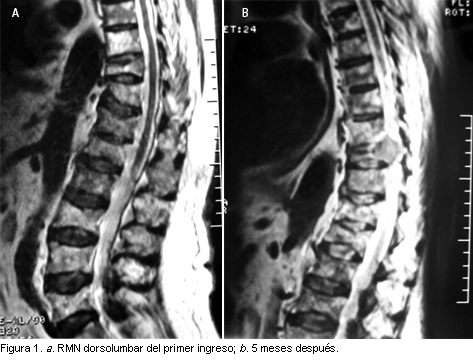
Se realiza una resonancia magnética nuclear (RMN) de columna dorsolumbar en la que se observan acuñamientos múltiples de origen osteopénico y afección del cuerpo y pedículo derecho de D10. No se visualiza masa paravertebral ni afección intrarraquídea (figura 1a). En la gammagrafía ósea existen diversos focos de hipercaptación a nivel de hombro derecho, arco costal derecho y columna dorsolumbar no sugestivos de metástasis.
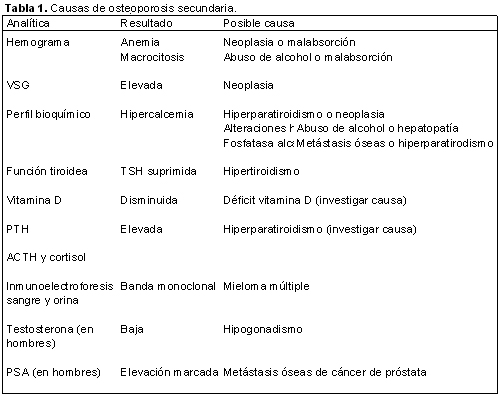
Ante la sospecha de osteoporosis grave difusa de origen secundario, se solicitan determinaciones hormonales para descartar su origen patológico; los resultados son normales (PTH: 33.8 pg/ml; gammagrafía y SPECT de paratiroides normales, TSH: 1 220 mU/l; ACTH: 21 pg/ml; cortisol a las 8 horas: 16.2 nmol/l), por lo que el paciente es dado de alta con diagnóstico de osteoporosis idiopática.
Cinco meses más tarde el sujeto ingresa nuevamente por dolor óseo generalizado progresivo e incapacitante. A la exploración, el paciente presenta afección del estado general, intenso dolor a la movilización de tórax y extremidades, tanto superiores como inferiores, y acentuación de la cifosis dorsal. Neurológicamente existe disminución discreta de fuerza (4/5) en extremidad inferior izquierda e hipoestesia táctil en extremidad inferior derecha. En el hemograma persiste la anemia normocítica (hemoglobina = 11.3 mg/dl) y, además, se observa un notable aumento de la VSG (42 mm). En la bioquímica se sigue manteniendo una buena función renal con cifras de urea y creatinina de 20 y 0.6 mg/dl, respectivamente, pero se detecta un aumento de los niveles de beta 2 microglobulina (2.47 mg/l) y de calcio (11.8 mg/dl). La hipercalcemia se considera iatrogénica, dada su corrección tras suprimir la administración de calcio y vitamina D. También existe un descenso generalizado de las inmunoglobulinas séricas (IgG: 487 mg/dl; IgA: 36 mg/dl; IgM: 29; IgE: 9 mg/dl); en la inmunoelectroforesis de sangre y orina no se detectan picos monoclonales ni cadenas ligeras. La PTH continúa en valores normales.
Se repiten las pruebas complementarias de imágenes, en la RMN de tórax se aprecia fractura de segundo arco costal derecho y múltiples imágenes osteolíticas en la escápula; en la RMN dorsolumbar, múltiples acuñamientos vertebrales ya conocidos, y una masa de partes blandas a nivel D10 que invade el canal (figura 1b).
Con la sospecha de mieloma múltiple como enfermedad de base, se realiza aspirado-biopsia de médula ósea en la que se halla un infiltrado de células plasmáticas superior al 20%, confirmándose el diagnóstico. El paciente es trasladado al servicio de hematología de nuestro hospital para recibir tratamiento con esteroides y quimioterapia.
Discusión
El mieloma múltiple, o enfermedad de Kahler, es una proliferación maligna de células plasmáticas, principalmente en la médula ósea, que se acompaña generalmente de un exceso de producción de una inmunoglobulina cualitativamente normal, homogénea y monoclonal o de una cadena ligera. No se asocia con factores etiológicos conocidos. La media de edad de presentación se sitúa en torno de los 70 años, es excepcional antes de los 40. Se caracteriza por presentar afección ósea (dolores óseos casi constantes, radiculalgias y fracturas patológicas, principalmente); insuficiencia renal crónica por tubulopatía, o aguda precipitada por factores desencadenantes como la toma de antiinflamatorios no esteroides (AINE) o la infusión de contraste yodado; anemia, aumento de la velocidad de sedimentación, proteinuria e hipercalcemia. Los síntomas iniciales más comunes son astenia, dolor óseo e infecciones recurrentes, sobre todo urinarias y pulmonares. Todos estos hallazgos se asocian a la presencia de un componente monoclonal elevado en sangre, en orina o en ambas, detectado por inmunoelectroforesis que, junto con la biopsia de médula ósea, permite el diagnóstico de certeza.
El componente monoclonal puede faltar, ya sea porque la cantidad de proteína monoclonal sea mínima (mielomas hiposecretores) o porque esté realmente ausente, como sucede tanto en los mielomas no secretores, como en los mielomas de cadenas ligeras.
El mieloma no secretor representa menos del 1% de todos los mielomas. Su peculiaridad se cree que es debida a la incapacidad para sintetizar o secretar el componente M por parte de las células plasmáticas malignas, aunque el mecanismo fisiopatológico exacto se desconoce.
Constituyen casos de difícil diagnóstico debido a la ausencia de las alteraciones analíticas típicas del mieloma secretor; debe sospechárselo ante la elevación de la proteína C-reactiva, la velocidad de sedimentación y la beta 2 microglobulina, así como ante la disminución de las inmunoglobulinas séricas. Las pruebas de imagen son muy útiles, no sólo para apoyar el diagnóstico ante la presencia de focos osteolíticos, sino también para evaluar la respuesta terapéutica, dado que no nos podemos apoyar en las variaciones cuantitativas del componente M.
Nuestro paciente inicialmente presentaba clínica dolorosa inespecífica con patrón radiológico de lesiones osteopénicas, sin alteraciones analíticas específicas que apoyasen la sospecha diagnóstica de mieloma. En sólo cinco meses sufrió empeoramiento clínico con importante repercusión en el estado general y la calidad de vida. En la analítica se halló un notable aumento de la VSG y de los niveles de beta 2 microglobulina con respecto a las cifras previas y hubo importantes cambios en imágenes.
En el manejo diagnóstico de la osteoporosis se debe incluir la identificación de causas secundarias potencialmente tratables, como hipogonadismo masculino, hiperparatiroidismo primario e hipertiroidismo. Para ello es imprescindible una correcta anamnesis y la realización de una serie de determinaciones analíticas sencillas que nos pueden ayudar a orientar el diagnóstico (tabla 1). Es importante incluir el mieloma múltiple entre las causas de osteoporosis secundaria, aunque la analítica no sea específica de mieloma. Recientemente, Abrahamsen y col. realizaron un estudio observacional retrospectivo sobre 799 pacientes con sospecha de osteoporosis; en 1 de cada 20 pacientes con osteoporosis confirmada el diagnóstico fue mieloma mútiple o gammapatía monoclonal de significado incierto. El estudio confirma la importancia de realizar a modo de pesquisa la determinación del componente M en suero y orina en todos los casos de osteoporosis. Evidentemente, el problema surge en los casos de mielomas múltiples no secretores, como el aquí presentado, en los que el componente M está ausente.
Los autores no manifiestan conflictos de intereses.
El mieloma no secretor representa menos del 1% de todos los mielomas. Clínicamente, hematológicamente y bioquímicamente los hallazgos son similares a los encontrados en el mieloma secretor. Su peculiaridad reside en la ausencia de componente monoclonal tanto en suero como en orina. El diagnóstico puede retrasarse dado que en forma inicial presenta un patrón clínico muy inespecífico, especialmente aquellos casos en que los estudios radiológicos no revelan lesiones líticas. Se presenta un caso de mieloma múltiple no secretor de difícil diagnóstico.
Caso clínico
Se trata de un varón de 72 años con antecedentes personales de diabetes mellitus tipo 2, traumatismo tras caída accidental con aplastamiento vertebral a nivel de L1 y fractura de cóccix secundaria, discopatía degenerativa y distimia.
Ingresa en nuestro servicio procedente de consultas externas, para diagnóstico y tratamiento de lumbociatalgia bilateral progresiva de predominio izquierdo asociada con cervicobraquialgia derecha, de aproximadamente 8 meses de evolución y de características inflamatorias. No presenta clínica sistémica. En la exploración física presenta un buen estado general, destaca un soplo sistólico en foco aórtico a la auscultación cardíaca y, neurológicamente, abolición de reflejos osteotendinosos aquíleos y patelares bilaterales e hipoestesia táctil en cara externa de pierna izquierda. No se observa déficit motor. En el tacto rectal: próstata de aspecto adenomatoso de grado II.
A su ingreso se le realiza una serie de pruebas complementarias básicas que incluyen perfil analítico general y radiografías. En el hemograma destaca una anemia normocítica leve (hemoglobina = 11 mg/dl) con una velocidad de sedimentación globular (VSG) de 13 mm. En la bioquímica (incluido el perfil hepático y lipídico) no existen hallazgos patológicos, presenta niveles de urea y creatinina de 19 mg/dl y 0.6 mg/dl, respectivamente, y calcemia de 9.52 mg/dl con fosfatemia de 4.46 mg/dl. El proteinograma muestra un descenso de proteínas (5.5 g/dl) a expensas de las gammaglobulinas (7.1%).
En la radiografía de tórax se observan signos de enfisema pulmonar y osteólisis en húmero derecho y ambas clavículas. En la radiografía simple de columna vertebral lumbar presenta osteoporosis difusa con acuñamientos vertebrales D9-D12 y patrón permeable L3-L5.
Se realiza una resonancia magnética nuclear (RMN) de columna dorsolumbar en la que se observan acuñamientos múltiples de origen osteopénico y afección del cuerpo y pedículo derecho de D10. No se visualiza masa paravertebral ni afección intrarraquídea (figura 1a). En la gammagrafía ósea existen diversos focos de hipercaptación a nivel de hombro derecho, arco costal derecho y columna dorsolumbar no sugestivos de metástasis.
Ante la sospecha de osteoporosis grave difusa de origen secundario, se solicitan determinaciones hormonales para descartar su origen patológico; los resultados son normales (PTH: 33.8 pg/ml; gammagrafía y SPECT de paratiroides normales, TSH: 1 220 mU/l; ACTH: 21 pg/ml; cortisol a las 8 horas: 16.2 nmol/l), por lo que el paciente es dado de alta con diagnóstico de osteoporosis idiopática.
Cinco meses más tarde el sujeto ingresa nuevamente por dolor óseo generalizado progresivo e incapacitante. A la exploración, el paciente presenta afección del estado general, intenso dolor a la movilización de tórax y extremidades, tanto superiores como inferiores, y acentuación de la cifosis dorsal. Neurológicamente existe disminución discreta de fuerza (4/5) en extremidad inferior izquierda e hipoestesia táctil en extremidad inferior derecha. En el hemograma persiste la anemia normocítica (hemoglobina = 11.3 mg/dl) y, además, se observa un notable aumento de la VSG (42 mm). En la bioquímica se sigue manteniendo una buena función renal con cifras de urea y creatinina de 20 y 0.6 mg/dl, respectivamente, pero se detecta un aumento de los niveles de beta 2 microglobulina (2.47 mg/l) y de calcio (11.8 mg/dl). La hipercalcemia se considera iatrogénica, dada su corrección tras suprimir la administración de calcio y vitamina D. También existe un descenso generalizado de las inmunoglobulinas séricas (IgG: 487 mg/dl; IgA: 36 mg/dl; IgM: 29; IgE: 9 mg/dl); en la inmunoelectroforesis de sangre y orina no se detectan picos monoclonales ni cadenas ligeras. La PTH continúa en valores normales.
Se repiten las pruebas complementarias de imágenes, en la RMN de tórax se aprecia fractura de segundo arco costal derecho y múltiples imágenes osteolíticas en la escápula; en la RMN dorsolumbar, múltiples acuñamientos vertebrales ya conocidos, y una masa de partes blandas a nivel D10 que invade el canal (figura 1b).
Con la sospecha de mieloma múltiple como enfermedad de base, se realiza aspirado-biopsia de médula ósea en la que se halla un infiltrado de células plasmáticas superior al 20%, confirmándose el diagnóstico. El paciente es trasladado al servicio de hematología de nuestro hospital para recibir tratamiento con esteroides y quimioterapia.
Discusión
El mieloma múltiple, o enfermedad de Kahler, es una proliferación maligna de células plasmáticas, principalmente en la médula ósea, que se acompaña generalmente de un exceso de producción de una inmunoglobulina cualitativamente normal, homogénea y monoclonal o de una cadena ligera. No se asocia con factores etiológicos conocidos. La media de edad de presentación se sitúa en torno de los 70 años, es excepcional antes de los 40. Se caracteriza por presentar afección ósea (dolores óseos casi constantes, radiculalgias y fracturas patológicas, principalmente); insuficiencia renal crónica por tubulopatía, o aguda precipitada por factores desencadenantes como la toma de antiinflamatorios no esteroides (AINE) o la infusión de contraste yodado; anemia, aumento de la velocidad de sedimentación, proteinuria e hipercalcemia. Los síntomas iniciales más comunes son astenia, dolor óseo e infecciones recurrentes, sobre todo urinarias y pulmonares. Todos estos hallazgos se asocian a la presencia de un componente monoclonal elevado en sangre, en orina o en ambas, detectado por inmunoelectroforesis que, junto con la biopsia de médula ósea, permite el diagnóstico de certeza.
El componente monoclonal puede faltar, ya sea porque la cantidad de proteína monoclonal sea mínima (mielomas hiposecretores) o porque esté realmente ausente, como sucede tanto en los mielomas no secretores, como en los mielomas de cadenas ligeras.
El mieloma no secretor representa menos del 1% de todos los mielomas. Su peculiaridad se cree que es debida a la incapacidad para sintetizar o secretar el componente M por parte de las células plasmáticas malignas, aunque el mecanismo fisiopatológico exacto se desconoce.
Constituyen casos de difícil diagnóstico debido a la ausencia de las alteraciones analíticas típicas del mieloma secretor; debe sospechárselo ante la elevación de la proteína C-reactiva, la velocidad de sedimentación y la beta 2 microglobulina, así como ante la disminución de las inmunoglobulinas séricas. Las pruebas de imagen son muy útiles, no sólo para apoyar el diagnóstico ante la presencia de focos osteolíticos, sino también para evaluar la respuesta terapéutica, dado que no nos podemos apoyar en las variaciones cuantitativas del componente M.
Nuestro paciente inicialmente presentaba clínica dolorosa inespecífica con patrón radiológico de lesiones osteopénicas, sin alteraciones analíticas específicas que apoyasen la sospecha diagnóstica de mieloma. En sólo cinco meses sufrió empeoramiento clínico con importante repercusión en el estado general y la calidad de vida. En la analítica se halló un notable aumento de la VSG y de los niveles de beta 2 microglobulina con respecto a las cifras previas y hubo importantes cambios en imágenes.
En el manejo diagnóstico de la osteoporosis se debe incluir la identificación de causas secundarias potencialmente tratables, como hipogonadismo masculino, hiperparatiroidismo primario e hipertiroidismo. Para ello es imprescindible una correcta anamnesis y la realización de una serie de determinaciones analíticas sencillas que nos pueden ayudar a orientar el diagnóstico (tabla 1). Es importante incluir el mieloma múltiple entre las causas de osteoporosis secundaria, aunque la analítica no sea específica de mieloma. Recientemente, Abrahamsen y col. realizaron un estudio observacional retrospectivo sobre 799 pacientes con sospecha de osteoporosis; en 1 de cada 20 pacientes con osteoporosis confirmada el diagnóstico fue mieloma mútiple o gammapatía monoclonal de significado incierto. El estudio confirma la importancia de realizar a modo de pesquisa la determinación del componente M en suero y orina en todos los casos de osteoporosis. Evidentemente, el problema surge en los casos de mielomas múltiples no secretores, como el aquí presentado, en los que el componente M está ausente.
Los autores no manifiestan conflictos de intereses.
Vanesa Bernal Monterde, Hospital Clínico Universitario Lozano Blesa, 50008, Zaragoza, España,
e-mail: vbernalm@gmail.com
1. Confavreus C. Myélome múltiple des os. Institut La Conférence Hippocrate 2005;1-12.
2. Anderson KC et al. Multiple myeloma. Programa educacional. ASH 2002; 214-240.
3. Pinto F. Non-secretory myeloma síndrome. Resentí Prog Med 2001; 92(9):533-6.
4. Kyle RA, Rajkumar SV. Multiple myeloma. N Engl J Med 2004; 351:1860-73.
5. Dataille R, Harousseau JL. Multiple myeloma. N Engl J Med 1997; 336:1657-1664.
6. Tuck SP, Francis RM. Postgrad Med J 2002; 78:526-532.
7. Abrahamsen B, Andersen I, Christensen S, Skov J, Brixen K. Utility of testing for monoclonal bands in serum of patients with suspected osteoporosis: retrospective, cross sectional study. BMJ 2005; 330:818-822.
Está expresamente prohibida la redistribución y la redifusión de todo o parte de los
contenidos de la Sociedad Iberoamericana de Información Científica (SIIC) S.A. sin
previo y expreso consentimiento de SIIC.
ua91218

