

-
 Alergia
Alergia -
 Anestesiología
Anestesiología -
 Bioquímica
Bioquímica
-
 Cardiología
Cardiología
-
 Cirugía
Cirugía
-
 Dermatología
Dermatología
-
 Endocrinología y Metabolismo
Endocrinología y Metabolismo
-
 Enfermería
Enfermería
-
 Gastroenterología
Gastroenterología
-
 Hematología
Hematología
-
 Infectología
Infectología
-
 Inmunología
Inmunología
-
 Medicina Interna
Medicina Interna
-
 Nefrología
Nefrología
-
 Neumonología
Neumonología
-
 Neurología
Neurología
-
 Nutrición
Nutrición
-
 Obstetricia y Ginecología
Obstetricia y Ginecología
-
 Odontología
Odontología
-
 Oncología
Oncología
-
 Otorrinolaringología
Otorrinolaringología
-
 Pediatría
Pediatría
-
 Salud Mental
Salud Mental
-
 Salud Pública
Salud Pública
-
 Urología
Urología
-
 Más Especialidades
Más Especialidades
- Índice
-
Acceso por especialidad
-
Alergia
-
Anestesiología
-
Bioquímica
-
Cardiología
-
Cirugía
-
Dermatología
-
Endocrinología y Metabolismo
-
Enfermería
-
Gastroenterología
-
Hematología
-
Infectología
-
Inmunología
-
Medicina Interna
-
Nefrología
-
Neumonología
-
Neurología
-
Nutrición
-
Obstetricia y Ginecología
-
Odontología
-
Oncología
-
Otorrinolaringología
-
Pediatría
-
Salud Mental
-
Salud Pública
-
Urología
-
Más Especialidades
-
Noticias biomédicas
-
Fuentes informativas
- Editoriales
-
COVID-19
-
INFORMES CIENTÍFICOS
- Preguntas con Respuesta
- América Latina Investiga
- Completos revisados (full text)
-
NOTICIAS/OPINIONES
-
Expertos invitados
- Por especialidad
- de Iberoamérica
- del mundo
- Entrevistas
- Casos clínicos
-
Crónicas de Autores
- Authors’ Reports
-
Instrucciones para autores
-
Expertos Preguntan
-
Novedades
- ViASIIC
- Medicina del Dolor
-
Por especialidad
-
Iberomédica
-
Informes comentados
- Textos Completos Autorizados
- Preocupaciones Profesionales
-
Red Científica Iberoamericana (RedCIbe)
-
Conceptos Categoricos
-
Acceso por especialidad

-
Alergia
-
Anestesiología
-
Bioquímica
-
Cardiología
-
Cirugía
-
Dermatología
-
Endocrinología y Metabolismo
-
Enfermería
-
Gastroenterología
-
Hematología
-
Infectología
-
Inmunología
-
Medicina Interna
-
Nefrología
-
Neumonología
-
Neurología
-
Nutrición
-
Obstetricia y Ginecología
-
Odontología
-
Oncología
-
Otorrinolaringología
-
Pediatría
-
Salud Mental
-
Salud Pública
-
Urología
-
Más Especialidades
-
Noticias biomédicas
-
Fuentes informativas
- Editoriales
- Covid-19
-
Informes Científicos
- Preguntas con Respuesta
- América Latina Investiga
- Completos revisados (full text)
- Noticias/Opiniones
- Noticias (castellano/portugués)
- Noticias (otros idiomas)
- Expertos invitados
- Por especialidad
- de Iberoamérica
- del mundo
- Entrevistas
- Casos clínicos
-
Crónicas de Autores
- Authors’ Reports
-
Instrucciones para autores
-
Expertos Preguntan
- Novedades
- ViASIIC
- Medicina del Dolor
-
Por especialidad
-
Iberomédica
-
Informes comentados
- Textos Completos Autorizados
- Preocupaciones Profesionales
- Red Científica Iberoamericana
- Por materia
- Por fecha
-
Conceptos Categóricos

Casos Clínicos
HIPERTENSION PULMONAR ASOCIADA A LUPUS ERITEMATOSO SISTEMICO: REPORTE DE CASO
La asociación de hipertensión arterial pulmonar con lupus eritematoso sistémico es infrecuente. Presentamos el caso de una paciente puérpera con diagnóstico, sin tratamiento convencional, a quien debido a un agravamiento de su enfermedad se le diagnosticó hipertensión pulmonar clase III.
Coautores
Alvaro Ramirez Toncel* Jhair Martinez Obando*
Medico, Clínica Privada Independencia*
Alvaro Ramirez Toncel* Jhair Martinez Obando*
Medico, Clínica Privada Independencia*
Clasificación en siicsalud
Artículos originales > Expertos del Mundo >
página /dato/casiic.php/153083
Especialidades




Artículos originales > Expertos del Mundo >
página /dato/casiic.php/153083
Especialidades
Primera edición en siicsalud
15 de noviembre, 2016
15 de noviembre, 2016
HIPERTENSION PULMONAR ASOCIADA A LUPUS ERITEMATOSO SISTEMICO: REPORTE DE CASO
(especial para SIIC © Derechos reservados)
(especial para SIIC © Derechos reservados)
Introducción
La hipertensión pulmonar (HTP) es una complicación grave que se presenta infrecuentemente en pacientes con lupus eritematoso sistémico (LES) con una prevalencia del 3% al 4%;1,2 suele ser una manifestación tardía de la enfermedad y se relaciona con la gravedad de la enfermedad renal, el grado de actividad y la concentración de proteína C, su etiología puede ser multifactorial, incluyendo enfermedad pulmonar intersticial y tromboembolismo pulmonar,3 y se la reconoce como predictor independiente de mortalidad.4 El fenómeno de Raynaud se presenta en el 75% de los pacientes que asocian la HTP con LES.5 Entre los anticuerpos asociados con la presencia de HTP y LES se mencionan los antifosfolípidos, los anti-antígenos nucleares extraíbles anti-Smith (anti-Sm) y los anti-La/SSB. Como tratamiento farmacológico se incluyen antagonistas de los canales de calcio, prostaciclinas, antagonistas de los receptores de la endotelina, inhibidores de la fosfodiesterasa, corticosteroides e inmunosupesores.6-11
Caso clínico
Paciente de sexo femenino, de 25 años de edad, con antecedente de lupus eritematoso sistémico de un año de evolución en tratamiento homeopático, cursando puerperio de un mes consultó por disnea clase funcional III, por lo que se internó. Se constató como dato edemas generalizados con predominio de miembros inferiores, derrame pleural derecho y ascitis; fenómeno de Reynaud negativo. Con diagnóstico de crisis lúpica se inició tratamiento con metilprednisolona 1 g/día. Como datos de laboratorio relevantes se obtuvo TGO = 644 U/l, TGP = 235U/l, fosfatasa alcalina = 419 UI/l, bilirrubina total de 2.3 mg% (directa de 1.5 mg%), tiempo de protrombina 29%, RIN = 2.35; albuminemia = 2.1 g%, proteinuria de 1 g en 24 horas; proteína C-reactiva = 48 mg/l (valor normal hasta 6 mg/l), anticuerpos antinucleares positivo 1/320 (moteado fino), anticuerpos anticitoplasmáticos positivo 1/640 (granular), anticuerpos anti-antígeno nuclear extraíble Ro/SSA (anti-Ro/SSA) positivo, anticuerpo anti-LA/SSB positivo, anticuerpo anti-Sm positivo débil, anticuerpos anticentrómero negativo, anticuerpos anti-Scl 70 negativo, anticuerpo anti-ADN nativo negativo, anticuerpos anticardiolipina IgG e IgM negativos, anticuerpos antimieloperoxidasa negativo, anticuerpos antiproteasa negativos, anticuerpos antineutrófilos negativo. En la radiogafía de tórax se observa agrandamiento de la arteria pulmonar (Figura 1).

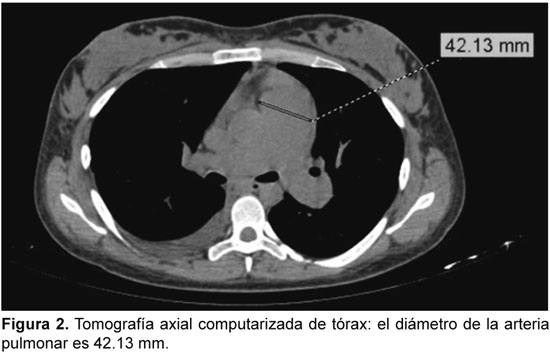
En el ecocardiograma se observan dimensiones y función de ventrículo izquierdo normales, dilatación de cavidades derechas, movimiento anormal del septum, signos de congestión venosa sistémica, presión sistólica de arteria pulmonar de 63 mm Hg. En la ecografía abdominal, moderada cantidad de líquido libre perihepático, periesplénico, interasas y fondo de saco de Douglas. La angiotomografía de tórax con protocolo para embolismo pulmonar descartó dicho diagnóstico e informó tronco de la arteria pulmonar de 42.13 mm.
La paciente evolucionó favorablemente luego del tratamiento con metilprednisolona 1 gramo por 5 días, continuando con prednisona 60 mg/día, sildenafil 100 mg/día y diuréticos de asa. A los 23 días de ingreso se decidió efectuar cateterismo y prueba de óxido nítrico para definir la conducta definitiva; se constató presión media de aurícula derecha de 21 mm Hg, presión de ventrículo derecho de 83 mm Hg, presión de fin de diástole de 18 mm Hg, presiones de arteria pulmonar de 83, 50 y 63 mm Hg de sistólica, diastólica y media, respectivamente, presión de oclusión pulmonar de 6 mm Hg. Durante la administración de óxido nítrico la paciente presentó asistolia refractaria al tratamiento.
Discusión
Se define hipertensión pulmonar cuando la presión de la arteria pulmonar es mayor de 25 mm Hg registrada por cateterismo cardíaco derecho y en reposo.12 La HTP puede presentarse en forma primaria o ser secundaria a otras enfermedades, entre ellas en la actualidad se consideran las enfermedades del colágeno como una causa importante de su manifestación, siendo una de las principales causas de disnea de estos pacientes, puede presentarse con afectación del intersticio pulmonar o sin ella, la evolución de la colagenopatía es la responsable del incremento de la HTP,13 e incide directamente en la morbimortalidad. Entre las colagenopatías, la que se asocia con mayor frecuencia a HTP es la esclerodermia, su asociación con el lupus eritematoso sistémico es poco frecuente.13,14 Entre los mecanismos fisiopatológicos implicados en la patogénesis de la HTP en las colagenopatías figuran la vasoconstricción pulmonar hipóxica con remodelamiento vascular, obstrucción vascular, fibrosis perivascular, inflamación vascular, embolia pulmonar, disfunción endotelial, disfunción del músculo liso vascular; particularmente en esclerodermia y síndrome CREST (calcinosis, fenómeno de Raynaud, trastornos de la motilidad esofágica, esclerodactilia, telangiectasias) se han descrito procesos autoinmunitarios contra el endotelio e hipertensión pulmonar pasiva debida a disfunción diastólica.
El LES se caracteriza por la presencia de autoanticuerpos contra antígenos nuclear, puede generar disfunción de cualquier órgano de la economía; respecto del aparato respiratorio, puede afectar cualquiera de sus componentes y puede manifestarse como pleuritis, derrame pleural, neumonitis aguda, hemorragia pulmonar, enfermedad intersticial difusa, hipoxemia reversible e hipertensión pulmonar.15 Se han descrito factores distintivos de los pacientes con HTP y LES: el 75% presenta fenómeno de Raynaud, en comparación con el 25% al 40% que no lo presentan, los anticuerpos antifosfolípidos se presentan hasta en el 68% de los casos y también se menciona la presencia de los anti-Sm y anti-La/SSB.
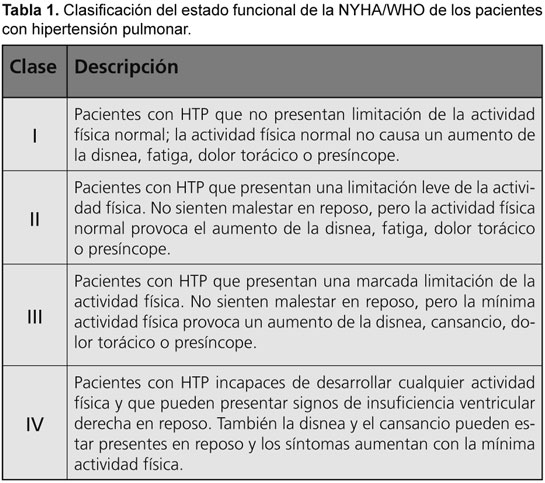
La supervivencia de los pacientes con hipertensión pulmonar sin tratamiento, de acuerdo con la clasificación funcional de la Organización Mundial de la Salud (Tabla 1) es de seis años para los que se encuentran en clase funcional (CF) I/II, 2.5 años para los de CF III y de seis meses para los de CF IV.16 Se han descrito factores predictivos de mala evolución en pacientes con HTP, entre ellos caben destacar la edad mayor de 60 años y el sexo masculino,17 el desplazamiento en sístole y el índice de performance miocárdica del ventrículo derecho,18-20 dentro de los parámetros hemodinámicos marcadores de mal pronóstico se citan el índice cardíaco, la saturación venosa mixta, la resistencia vascular pulmonar y la presión en la aurícula derecha.16 La disfunción ventricular derecha con presión de fin de diástole mayor de 20 mm Hg se asocia con arritmias y muerte súbita;21 una desaturación de oxígeno mayor del 10% en la prueba de marcha de seis minutos es índice de gravedad,22 mientras que los pacientes que en dicha prueba toleran caminar 330 metros o más tienen mayor supervivencia.23
En cuanto a los marcadores bioquímicos, los valores de propéptido natriurético cerebral N-terminal (Nt-proBNP) por encma de 1400 pg/ml se asocian con mayor mortalidad.24 El tratamiento de la HTP debe individualizarse según la clase funcional y su etiología; en los últimos años se ha producido un importante avance sobre los principales trastornos: la vasoconstricción, el remodelamiento de la pared vascular y la trombosis. No se ha demostrado si la actividad física prolonga la supervivencia de estos pacientes;25,26 no obstante, se recomienda entrenamiento especializado en pacientes con enfermedades cardiovasculares o respiratorias graves. La causa de la hipoxia en pacientes con HTP es debida a varios factores; no está demostrado que la oxigenoterapia crónica domiciliaria sea beneficiosa.
Se recomienda el uso de digoxina en casos de insuficiencia cardíaca y fibrilación auricular.27 Los diuréticos están indicados en pacientes con sobrecarga hídrica. Los anticoagulantes orales se indican a aquellos pacientes con hipertensión pulmonar tromboembólica crónica e hipertensión pulmonar idiopática.28,29 Los bloqueantes cálcicos se usan en el tratamiento crónico en los pacientes que responden a la prueba aguda de vasorreactividad,30 los más utilizados son la nifedipina, el diltiazem y la amlodipina; sin embargo, el 50% de los enfermos dejarán de responder dentro del año de tratamiento.31 En cuanto a los prostanoides, el epoprostenol mejora los síntomas, la capacidad al ejercicio y los parámetros hemodinámicos, se administra mediante un catéter venoso central; el iloprost es un análogo de la prostaciclina y su administración puede ser oral, intravenosa o en aerosol; el treprostinil puede administrarse en forma inhalatoria, intravenosa o subcutánea. El bosentán actúa inhibiendo los receptores de endotelina y mejora la capacidad al ejercicio, la clase funcional y las variables ecocardiográficas;32,33 el ambrisentán demostró mejorar la clase funcional y hemodinámica.34 Los inhibidores de la fosfodiesterasa como el sildenafil y el tadalafilo mejoran la capacidad al ejercicio y la clase funcional.35,36 En cuanto al tratamiento con glucocorticoides o inmnosupresores o ambos, se han presentado series de pacientes en los que se han encontrado resultados favorables en individuos tratados con ciclofosfamida sola o asociada con glucocorticoides en estadios iniciales de hipertensión pulmonar.37-39 En cuanto al tratamiento quirúrgico, la septostomía auricular consiste en realizar un cortocircuito de derecha a izquierda a nivel del foramen oval, con lo que se consigue disminuir la presión de la aurícula derecha, descomprimir el ventrículo derecho y aumentar la precarga izquierda, mejorando así el gasto cardíaco y el transporte de oxígeno; el trasplante pulmonar bilateral o el cardiopulmonar es una opción ante el fracaso en algunos pacientes seleccionados.
Conclusiones
Se presenta el caso clínico debido a la baja frecuencia de hipertensión de la arteria pulmonar asociada con lupus eritematoso sistémico. La paciente en estudio presentaba como anticuerpos descritos en lupus eritematoso sistémico asociados a hipertensión pulmonar anti-La/SSB y anti-SM, cabe destacar la ausencia de fenómeno de Raynaud, que suele registrarse en el 75% de los casos que presentan la asociación en LES y HTP. Por otro lado, la paciente, portadora de hipertensión pulmonar, cursó un embarazo sin tratamiento adecuado, cuando es conocida la alta mortalidad asociada (y contraindicada por algunos autores) incrementada por la falta de soporte hemodinámico.
La hipertensión pulmonar (HTP) es una complicación grave que se presenta infrecuentemente en pacientes con lupus eritematoso sistémico (LES) con una prevalencia del 3% al 4%;1,2 suele ser una manifestación tardía de la enfermedad y se relaciona con la gravedad de la enfermedad renal, el grado de actividad y la concentración de proteína C, su etiología puede ser multifactorial, incluyendo enfermedad pulmonar intersticial y tromboembolismo pulmonar,3 y se la reconoce como predictor independiente de mortalidad.4 El fenómeno de Raynaud se presenta en el 75% de los pacientes que asocian la HTP con LES.5 Entre los anticuerpos asociados con la presencia de HTP y LES se mencionan los antifosfolípidos, los anti-antígenos nucleares extraíbles anti-Smith (anti-Sm) y los anti-La/SSB. Como tratamiento farmacológico se incluyen antagonistas de los canales de calcio, prostaciclinas, antagonistas de los receptores de la endotelina, inhibidores de la fosfodiesterasa, corticosteroides e inmunosupesores.6-11
Caso clínico
Paciente de sexo femenino, de 25 años de edad, con antecedente de lupus eritematoso sistémico de un año de evolución en tratamiento homeopático, cursando puerperio de un mes consultó por disnea clase funcional III, por lo que se internó. Se constató como dato edemas generalizados con predominio de miembros inferiores, derrame pleural derecho y ascitis; fenómeno de Reynaud negativo. Con diagnóstico de crisis lúpica se inició tratamiento con metilprednisolona 1 g/día. Como datos de laboratorio relevantes se obtuvo TGO = 644 U/l, TGP = 235U/l, fosfatasa alcalina = 419 UI/l, bilirrubina total de 2.3 mg% (directa de 1.5 mg%), tiempo de protrombina 29%, RIN = 2.35; albuminemia = 2.1 g%, proteinuria de 1 g en 24 horas; proteína C-reactiva = 48 mg/l (valor normal hasta 6 mg/l), anticuerpos antinucleares positivo 1/320 (moteado fino), anticuerpos anticitoplasmáticos positivo 1/640 (granular), anticuerpos anti-antígeno nuclear extraíble Ro/SSA (anti-Ro/SSA) positivo, anticuerpo anti-LA/SSB positivo, anticuerpo anti-Sm positivo débil, anticuerpos anticentrómero negativo, anticuerpos anti-Scl 70 negativo, anticuerpo anti-ADN nativo negativo, anticuerpos anticardiolipina IgG e IgM negativos, anticuerpos antimieloperoxidasa negativo, anticuerpos antiproteasa negativos, anticuerpos antineutrófilos negativo. En la radiogafía de tórax se observa agrandamiento de la arteria pulmonar (Figura 1).
En el ecocardiograma se observan dimensiones y función de ventrículo izquierdo normales, dilatación de cavidades derechas, movimiento anormal del septum, signos de congestión venosa sistémica, presión sistólica de arteria pulmonar de 63 mm Hg. En la ecografía abdominal, moderada cantidad de líquido libre perihepático, periesplénico, interasas y fondo de saco de Douglas. La angiotomografía de tórax con protocolo para embolismo pulmonar descartó dicho diagnóstico e informó tronco de la arteria pulmonar de 42.13 mm.
La paciente evolucionó favorablemente luego del tratamiento con metilprednisolona 1 gramo por 5 días, continuando con prednisona 60 mg/día, sildenafil 100 mg/día y diuréticos de asa. A los 23 días de ingreso se decidió efectuar cateterismo y prueba de óxido nítrico para definir la conducta definitiva; se constató presión media de aurícula derecha de 21 mm Hg, presión de ventrículo derecho de 83 mm Hg, presión de fin de diástole de 18 mm Hg, presiones de arteria pulmonar de 83, 50 y 63 mm Hg de sistólica, diastólica y media, respectivamente, presión de oclusión pulmonar de 6 mm Hg. Durante la administración de óxido nítrico la paciente presentó asistolia refractaria al tratamiento.
Discusión
Se define hipertensión pulmonar cuando la presión de la arteria pulmonar es mayor de 25 mm Hg registrada por cateterismo cardíaco derecho y en reposo.12 La HTP puede presentarse en forma primaria o ser secundaria a otras enfermedades, entre ellas en la actualidad se consideran las enfermedades del colágeno como una causa importante de su manifestación, siendo una de las principales causas de disnea de estos pacientes, puede presentarse con afectación del intersticio pulmonar o sin ella, la evolución de la colagenopatía es la responsable del incremento de la HTP,13 e incide directamente en la morbimortalidad. Entre las colagenopatías, la que se asocia con mayor frecuencia a HTP es la esclerodermia, su asociación con el lupus eritematoso sistémico es poco frecuente.13,14 Entre los mecanismos fisiopatológicos implicados en la patogénesis de la HTP en las colagenopatías figuran la vasoconstricción pulmonar hipóxica con remodelamiento vascular, obstrucción vascular, fibrosis perivascular, inflamación vascular, embolia pulmonar, disfunción endotelial, disfunción del músculo liso vascular; particularmente en esclerodermia y síndrome CREST (calcinosis, fenómeno de Raynaud, trastornos de la motilidad esofágica, esclerodactilia, telangiectasias) se han descrito procesos autoinmunitarios contra el endotelio e hipertensión pulmonar pasiva debida a disfunción diastólica.
El LES se caracteriza por la presencia de autoanticuerpos contra antígenos nuclear, puede generar disfunción de cualquier órgano de la economía; respecto del aparato respiratorio, puede afectar cualquiera de sus componentes y puede manifestarse como pleuritis, derrame pleural, neumonitis aguda, hemorragia pulmonar, enfermedad intersticial difusa, hipoxemia reversible e hipertensión pulmonar.15 Se han descrito factores distintivos de los pacientes con HTP y LES: el 75% presenta fenómeno de Raynaud, en comparación con el 25% al 40% que no lo presentan, los anticuerpos antifosfolípidos se presentan hasta en el 68% de los casos y también se menciona la presencia de los anti-Sm y anti-La/SSB.
La supervivencia de los pacientes con hipertensión pulmonar sin tratamiento, de acuerdo con la clasificación funcional de la Organización Mundial de la Salud (Tabla 1) es de seis años para los que se encuentran en clase funcional (CF) I/II, 2.5 años para los de CF III y de seis meses para los de CF IV.16 Se han descrito factores predictivos de mala evolución en pacientes con HTP, entre ellos caben destacar la edad mayor de 60 años y el sexo masculino,17 el desplazamiento en sístole y el índice de performance miocárdica del ventrículo derecho,18-20 dentro de los parámetros hemodinámicos marcadores de mal pronóstico se citan el índice cardíaco, la saturación venosa mixta, la resistencia vascular pulmonar y la presión en la aurícula derecha.16 La disfunción ventricular derecha con presión de fin de diástole mayor de 20 mm Hg se asocia con arritmias y muerte súbita;21 una desaturación de oxígeno mayor del 10% en la prueba de marcha de seis minutos es índice de gravedad,22 mientras que los pacientes que en dicha prueba toleran caminar 330 metros o más tienen mayor supervivencia.23
En cuanto a los marcadores bioquímicos, los valores de propéptido natriurético cerebral N-terminal (Nt-proBNP) por encma de 1400 pg/ml se asocian con mayor mortalidad.24 El tratamiento de la HTP debe individualizarse según la clase funcional y su etiología; en los últimos años se ha producido un importante avance sobre los principales trastornos: la vasoconstricción, el remodelamiento de la pared vascular y la trombosis. No se ha demostrado si la actividad física prolonga la supervivencia de estos pacientes;25,26 no obstante, se recomienda entrenamiento especializado en pacientes con enfermedades cardiovasculares o respiratorias graves. La causa de la hipoxia en pacientes con HTP es debida a varios factores; no está demostrado que la oxigenoterapia crónica domiciliaria sea beneficiosa.
Se recomienda el uso de digoxina en casos de insuficiencia cardíaca y fibrilación auricular.27 Los diuréticos están indicados en pacientes con sobrecarga hídrica. Los anticoagulantes orales se indican a aquellos pacientes con hipertensión pulmonar tromboembólica crónica e hipertensión pulmonar idiopática.28,29 Los bloqueantes cálcicos se usan en el tratamiento crónico en los pacientes que responden a la prueba aguda de vasorreactividad,30 los más utilizados son la nifedipina, el diltiazem y la amlodipina; sin embargo, el 50% de los enfermos dejarán de responder dentro del año de tratamiento.31 En cuanto a los prostanoides, el epoprostenol mejora los síntomas, la capacidad al ejercicio y los parámetros hemodinámicos, se administra mediante un catéter venoso central; el iloprost es un análogo de la prostaciclina y su administración puede ser oral, intravenosa o en aerosol; el treprostinil puede administrarse en forma inhalatoria, intravenosa o subcutánea. El bosentán actúa inhibiendo los receptores de endotelina y mejora la capacidad al ejercicio, la clase funcional y las variables ecocardiográficas;32,33 el ambrisentán demostró mejorar la clase funcional y hemodinámica.34 Los inhibidores de la fosfodiesterasa como el sildenafil y el tadalafilo mejoran la capacidad al ejercicio y la clase funcional.35,36 En cuanto al tratamiento con glucocorticoides o inmnosupresores o ambos, se han presentado series de pacientes en los que se han encontrado resultados favorables en individuos tratados con ciclofosfamida sola o asociada con glucocorticoides en estadios iniciales de hipertensión pulmonar.37-39 En cuanto al tratamiento quirúrgico, la septostomía auricular consiste en realizar un cortocircuito de derecha a izquierda a nivel del foramen oval, con lo que se consigue disminuir la presión de la aurícula derecha, descomprimir el ventrículo derecho y aumentar la precarga izquierda, mejorando así el gasto cardíaco y el transporte de oxígeno; el trasplante pulmonar bilateral o el cardiopulmonar es una opción ante el fracaso en algunos pacientes seleccionados.
Conclusiones
Se presenta el caso clínico debido a la baja frecuencia de hipertensión de la arteria pulmonar asociada con lupus eritematoso sistémico. La paciente en estudio presentaba como anticuerpos descritos en lupus eritematoso sistémico asociados a hipertensión pulmonar anti-La/SSB y anti-SM, cabe destacar la ausencia de fenómeno de Raynaud, que suele registrarse en el 75% de los casos que presentan la asociación en LES y HTP. Por otro lado, la paciente, portadora de hipertensión pulmonar, cursó un embarazo sin tratamiento adecuado, cuando es conocida la alta mortalidad asociada (y contraindicada por algunos autores) incrementada por la falta de soporte hemodinámico.
Fernando Ricard Racca Velasquez, Munro. Vicente Lopez, Argentina,
e-mail: fracca@intramed.net
1. Pan TL, Thumboo J, Boey ML, primary and secondary hypertension in systemic lupus erythematous. Lupus 9:338-40, 2000.
2. Chung SM, Lee CK, Lee EY, et al. Clinical aspects of pulmonary hypertension y patients with idiopathic pulmonary arterial hypertension. Clin Rheumatol 25:866-72, 2006.
3. Prabu A, Gordon C. Pulmonary arterial hypertension in SLE: what do we know? Lupus 22:1274-85, 2013.
4. Min H, Lee J, Jung S, et al. Pulmonary hypertension in systemic lupus erythematosus: an independent predictor of patient survival. Korean J Intern Med 30(2):232-41, 2015.
5. Asherson RA, Higenbottam TW, Dinh Xuan AT, Khamashta MA, Hughes GR. Pulmonary hypertension in a lupus clinic: Experience with a twenty-four patients. J Rheumatol 17:1292-1298, 1990.
6. Robbins IM, Gaine SP, Schilz R, Tapson VF, Rubin LJ, Loyd JE. Epoprostenol for treatment of pulmonary Hypertension in patients with systemic lupus erythematous. Chest 117:14-8, 2000.
7. Rubin LJ, Badesch DB, Barst RJ, Galie N, Black CM, Keogha A. Bosentan therapy for pulmonary arterial hypertension. N Engl J Med 896-903, 2002.
8. Gonzalez-López L, Cardona Muñoz EG, Celis A, et al. Therappy with intermittent pulse cyclophosphamide for pulmonary hypertension associated with systemic lupus erythematous. Lupus 105-12, 2004.
9. Cozzi F, Montisci R, Marotta H, Bobbo F, Durigon N, Ruscazio M, et al. Bosentan therapy of pulmonary arterial hypertension in connective tissue diseases. Eur J Clin Invest 36(suppl 3):49-53, 2006.
10. Barst RJ, Langleben D, Frost A, Horn EM, Oudiz R, Shapiro S, et al. Sitaxsentan therapy for pulmonary arterial hypertension. Am J Respir Crit Care Med 169:441-447, 2004.
11. Wilkins MR, Paul GA, Strange JW, Tunariu N, Gin-Sing W, Banya WA, et al. Sildenafil Versus Endothelin Receptor Antagonist for Pulmonary Hypertension (SERAPH) study. Am J Respir Crit Care Med 171:1292-1297, 2005.
12. Sociedad Argentina de Cardiología, Asociación Argentina de Medicina Respiratoria, Sociedad Argentina de Reumatología. Consenso para el diagnóstico y tratamiento de la hipertensión de la arteria pulmonar. Revista Argentina de Cardiología 79(Supl 2), 2011.
13. McLaughlin V, McGoon M. Pulmonary Arterial Hypertension. Circulation 114:1417-31, 2006.
14. Tamborrini G, Distler O. Update in pulmonary hypertension associated with connective tissue diseases - a systematic literature review. Dtsch Med Wochenschr 133(Suppl 6):S199- 202, 2008.
15. Quismorio FP. Pulmonary manifestations of systemic lupus erythematous: in Walace D, Bebvra Hannahs H: Dubois' Lupus Erythematosus. Baltimore: Williams & Wilkins, 5th edition, pp. 673-92, 1997.
16. D´Alonso GE, Barst RJ, Yres SM, et al. Survival in patients with primary pulmonary. Results from a national prospective registry. Ann Intern Med 115:343-9, 1991.
17. Benza RL, Miller DP, Gomberg-Maitland, et al. Predicting survival in pulmonary arterial hypertension. Circulation 122:164-72, 2010.
18. Forfia PR, Fisher MR, Mathai SC, et al. Tricuspid annular displacement predicts survival in pulmonary hypertension. Am J Respir Crit Care Med 174:1034-41, 2006.
19. Tei C, Dujardin KS, Hodge DO, et al. Doppler echocardiographic index for assessment of global right ventricular function. J Am Soc Echocardiogr 838-47, 1996.
20. Kakouros N, Kakouros S, Lekakis J, et al. Tissue Doppler imaging of the tricuspid annulus and myocardical performance index in the evaluation of right ventricular involvement in the acute and late phase of a first inferior myocardical infarction. Echocardiography 28:311-9, 2011.
21. Guillinta P, Peterson KL, Yehuda O. Cardiac catheterization techniques in pulmonary hypertension. Cardiol Clin 22:401-15, 2004.
22. Paciocco G, Martinez F, Bossone E et al. Oxygen desaturationon the six minute walk test and mortality in untreated primary pulmonary hypertension. Eur Respir J 17:647-52, 2001.
23. Miyamoto S, Nagaya N, Satoh T, et al. Clinical correlates and prognostic significance of six minute walk test in patients with primary pulmonary hypertension: comparision with cardiopulmonary exercise esting. Am J Respir Crit Care Med 161:487-92, 2000.
24. Fijalkowska A, Kurzyna M, Torbicki A, et al. Serum N terminalbrain natriuretic peptide as a prognostic parameter in patients with pulmonary hypertension. Chest 129:1313-21, 2006.
25. Badesh DB, Champion HC, Sanchez MA, et al. Diagnosis and assessment of pulmonary arterial hypertension. J Am Coll Cardiol 54S55-S66, 2009.
26. Mereles D, Ehlken N, Kreuscher S, et al. Exercise and respiratory training improve exercise capacity and quality of life in patients with svere chronic pulmonary hypertension. Circulation 114:1482-9, 2006.
27. McLaughlin VV, Archer SL, Badesch DB, Barst RJ, Farber HW, Lindner JR, et al; American College of Cardiology Foundation Task Force on Expert Consensus Documents; American Heart Association; American College of Chest Physicians; American Thoracic Society, Inc; Pulmonary Hypertension Association. ACCF/AHA 2009 expert consensus document on pulmonary hypertension a report of the American College of Cardiology Foundation Task Force on Expert Consensus Documents and the American Heart Association developed in collaboration with the American College of Chest Physicians; American Thoracic Society, Inc.; and the Pulmonary Hypertension Association. J Am Coll Cardiol 53:1573-619, 2009.
28. Rich S, Kaufmann E, Levy PS. The effect of high doses of calciumchannel blockers on survival in primary pulmonary hypertension. N Engl J Med 327:76-81, 1992.
29. Fuster V, Steele PM, Edwards WD, Gersh BJ, McGoon MD, Frye RL. Primary pulmonary hypertension: natural history and the importance of thrombosis Circulation 70:580-7, 1984.
30. Sitbon O, Humbert M, Jais X, Ioos V, Hamid AM, Provencher S, et al. Long-term response to calcium channel blockers in idiopathic pulmonary arterial hypertension. Circulation 111:3105-11, 2005.
31. Lee SH, Rubin LJ. Current treatment strategies for pulmonary arterial hypertension. J Intern Med 258:199-215, 2005.
32. Galie N, Rubin L, Hoeper M, Jansa P, Al Hiti H, Meyer G, et al. Treatment of patients with mildly symptomatic pulmonary arterial hypertension with bosentan (EARLY study): a double-blind, randomised controlled trial. Lancet 371:2093-100, 2008.
33. Galie N, Beghetti M, Gatzoulis MA, Granton J, Berger RM, Lauer A, et al; Bosentan Randomized Trial of Endothelin Antagonist Therapy-5 (BREATHE-5) Investigators. Bosentan therapy in patients with Eisenmenger syndrome: a multicenter, double-blind, randomized, placebo-controlled study. Circulation 114:48-54, 2006.
34. Galie N, Olschewski H, Oudiz RJ, et al; Ambrisentan in Pulmonary Arterial Hypertension, Randomized, Double-Blind, Placebo-Controlled, Multicenter, Efficacy Studies (ARIES) Group. Ambrisentan for the treatment of pulmonary arterial hypertension: results of the ambrisentan in pulmonary arterial hypertension, randomized, double-blind, placebo-controlled, multicenter, efficacy (ARIES) study 1 and 2. Circulation 117:3010-9, 2008.
35. Galiè N, Ghofrani HA, Torbicki A, et al. Sildenafil citrate therapy for pulmonary arterial hypertension. N Engl J Med 353:2148-57, 2005.
36. Galiè N, Brundage BH, Ghofrani HA, et al; Pulmonary Arterial Hypertension and Response to Tadalafil (PHIRST) Study Group. Tadalafil therapy for pulmonary arterial hypertension. Circulation 119:2894-903, 2009.
37. Jais X, Launay D, Yaici A, Le Pavec J, Tchérakian C, Sitbon O et al. Immunosuppressive therapy in lupus- and mixed connective tissue disease-associated pulmonary arterial hypertension: a retrospective analysis of twenty-three cases. Arthritis Rheum 58:521-31, 2008.
37. Galiè N, Hoeper MM, Humbert M, et al; ESC Committee for Practice Guidelines (CPG). Guidelines for the diagnosis and treatment of pulmonary hypertension: The Task Force for the Diagnosis and Treatment of Pulmonary Hypertension of the European Society of Cardiology (ESC) and the European Respiratory Society (ERS), endorsed by the International Society of Heart and Lung Transplantation (ISHLT). Eur Heart J 30:2493-537, 2009.
38. Tanaka E, Harigai M, Tanaka M, Kawaguchi Y, Hara M, Kamatani N. Pulmonary hypertension in systemic lupus erythematosus: evaluation of clinical characteristics and response to inmunosuppresive treatment. J Rheumatol 29(2)282-7, 2002.
39. Sanchez O, Sitbon O, Jaïs X, Simanneau G, Humbert M. Immunosuppressive therapy in connective tissue diseases-associated pulmonary arterial hypertension. Chest 130(1):182-9, 2006.
Está expresamente prohibida la redistribución y la redifusión de todo o parte de los
contenidos de la Sociedad Iberoamericana de Información Científica (SIIC) S.A. sin
previo y expreso consentimiento de SIIC.
ua91218

